
Intrathecal interleukin-1β decreases sigma-1 receptor expression in spinal astrocytes in a murine model of neuropathic pain
By Sheu-Ran Choi, Ho Jae Han, Alvin J. Beitz, and Jang-Hern Lee
Excerpt from the article published in Biomedicine & Pharmacotherapy, Volume 144, December 2021, 112272, ISSN 0753-3322, published online October 1, 2021, DOI: https://doi.org/10.1016/j.biopha.2021.112272.
Editor’s Highlights
- The sigma-1 receptor (Sig-1R) is a unique ligand-operated intracellular receptor that is located on the mitochondria-associated endoplasmic reticulum (ER) membrane in astrocytes and neurons of the central and peripheral nervous systems.
- Sig-1R can translocate to the plasma membrane and interact with diverse ion channels and receptors contributing to the modulation of their activity and stability.
- Pathological activation of Sig-1R is involved in the pain abnormalities associated with neuropathic and inflammatory pain.
- Sig-1R in spinal cord astrocytes plays an important role in phosphorylation-mediated central sensitization, as well as, neuropathic pain development.
- Interleukin 1 beta (IL-1β) administration significantly reduces the development of neuropathic pain through the control of the spinal astrocyte Sig-1R expression.
Autors’ Highlights
- Intrathecal administration of IL-1β inhibits neuropathic pain development in CCI mice.
- IL-1β administration reduces the CCI-induced increased pGluN1 expression in the spinal cord.
- IL-1β administration inhibits the CCI-induced increased Sig-1R expression in spinal astrocytes.
- Stimulation of Sig-1R restores the inhibitory actions of IL-1β on mechanical allodynia and pGluN1 expression in CCI mice.
Abstract
The sigma-1 receptor (Sig-1R) plays an important role in spinal pain transmission by increasing phosphorylation of the N-methyl-D-aspartate (NMDA) receptor GluN1 subunit (pGluN1). As a result Sig-1R has been suggested as a novel therapeutic target for prevention of chronic pain. Here we investigated whether interleukin-1β (IL-1β) modulates the expression of the Sig-1R in spinal astrocytesduring the early phase of nerve injury, and whether this modulation affects spinal pGluN1 expression and the development of neuropathic pain following chronic constriction injury (CCI) of the sciatic nerve. Repeated intrathecal (i.t.) administration of IL-1β from days 0–3 post-surgery significantly reduced the increased pGluN1 expression at the Ser896 and Ser897 sites in the ipsilateral spinal cord, as well as, the development of mechanical allodynia and thermal hyperalgesiain the ipsilateral hind paw of CCI mice, which were restored by co-administration of IL-1 receptor antagonist with IL-1β. Sciatic nerve injury increased the expression of Sig-1R in astrocytes of the ipsilateral spinal cord, and this increase was suppressed by i.t. administration of IL-1β. Agonistic stimulation of the Sig-1R with PRE084 restored pGluN1 expression and the development of mechanical allodynia that were originally suppressed by IL-1β in CCI mice. Collectively these results demonstrate that IL-1β administration during the induction phase of neuropathic pain produces an analgesic effect on neuropathic pain development by controlling the expression of Sig-1R in spinal astrocytes.
1. Introduction
Not only neurons, but also glial cells play an important role in pain signaling transmission in the spinal cord [1], [2]. Glial cells synthesize and release a variety of cytokines, which transfer the signal to other adjacent glial cells and neurons modulating their activation [3]. Among these, interleukin-1β (IL-1β) is an important mediator of the inflammatory response and pain transmission in the peripheral and central nervous systems and it has been suggested to produce both nociceptive and antinociceptive effects [4], [5]. When administered into a variety of peripheral tissues, IL-1β has been shown to be nociceptive [5], [6], [7]. By contrast, it has been demonstrated that intrathecal (i.t.) administration of IL-1β suppresses thermal hyperalgesia in rats with peripheral inflammation [4]. It is consistent with our previous studies showing that early blockade of IL-1 receptor with i.t. administration of recombinant IL-1 receptor antagonist (IL-1ra) induces a significant enhancement of the development of mechanical allodynia and spinal astrocyte activation following carrageenan-induced inflammation [8] and chronic constriction injury (CCI)-induced peripheral neuropathy [9]. The IL-1 receptor is expressed in spinal astrocytes and IL-1β is transiently increased in the spinal cord during the early phase of pain following peripheral neuropathy [9], but it is not clear whether spinal IL-1β administration modulates the development of neuropathic pain and whether the changes in spinal astrocytes mediate this action of IL-1β.
The sigma-1 receptor (Sig-1R) is a unique ligand-operated intracellular receptor that is located on the mitochondria-associated endoplasmic reticulum (ER) membrane in astrocytes and neurons of the central and peripheral nervous systems[10]. Activated Sig-1Rs bind to IP3 receptors increasing Ca2+ efflux from the ER to the cytosol or mitochondria. Furthermore Sig-1R can translocate to the plasma membrane and interact with diverse ion channels and receptors contributing to the modulation of their activity and stability [11], [12]. Pathological activation of Sig-1R is involved in the pain abnormalities associated with neuropathic and inflammatory pain [13], [14]. In a previous study from our laboratories we demonstrated that sciatic nerve injury increased the expression of Sig-1R in spinal astrocytes during the induction phase of neuropathic pain, and blockade of this receptor during this period significantly suppressed spinal astrocyte activation and the development of mechanical allodynia [15]. In addition, Sig-1R increased protein kinase C (PKC)- and protein kinase A (PKA)-dependent phosphorylation of the N-methyl-D-aspartate (NMDA) receptor GluN1 subunit (pGluN1) [16], [17] contributing to the functional potentiation of NMDA receptors. Sig-1R in spinal cord astrocytes plays an important role in phosphorylation-mediated central sensitization, as well as, neuropathic pain development. However, it is not yet clear whether IL-1β modulates astrocyte Sig-1R expression in a murine model of neuropathic pain.
In this regard, the main purpose of the present study was to investigate whether exogenous IL-1β given during the induction phase of neuropathic pain modulates the expression of Sig-1R in spinal astrocytes, as well as, GluN1 phosphorylation in the spinal cord dorsal horn and to examine whether this modulation controls the development of neuropathic pain following peripheral nerve injury. Thus we investigated whether: (1) i.t. administration of IL-1β during the induction phase of neuropathic pain modulates neuropathic pain development and NMDA receptor phosphorylation in CCI mice, and if this modulation is reversed by blockade of the IL-1 receptor; (2) IL-1β administration modulates the expression of Sig-1R in spinal astrocytes of CCI mice; and (3) administration of the Sig-1R agonist, PRE084 restores the actions of IL-1β on spinal GluN1 phosphorylation and the development of neuropathic pain in a model of peripheral neuropathy.
…
3. Results
3.1. I.t. administration of IL-1β suppresses the CCI-induced development of neuropathic pain in the ipsilateral hind paw and this is blocked by co-administration of IL-1ra with IL-1β
In order to examine whether exogenous IL-1β modulates the CCI-induced development of neuropathic pain, we intrathecally administrated recombinant IL-1β (1, 3, and 10 ng) during the induction phase of neuropathic pain in CCI mice. Sciatic nerve injury increased the paw withdrawal response frequency (PWF, %) to non-noxious mechanical stimulation (mechanical allodynia) and decreased the paw withdrawal response latency (PWL, s) to noxious thermal stimulation (thermal hyperalgesia) in the ipsilateral hind paw at day 3 post-surgery (Fig. 1A and B; ** p < 0.01, *** p < 0.001 vs VEH-treated CCI group on day 0). Repeated i.t. administration of IL-1β from days 0–3 post-surgery dose-dependently suppressed the development of mechanical allodynia (Fig. 1A; F (7,40) = 5.218, p = 0.0003) and thermal hyperalgesia (Fig. 1B; F (7,40) = 11.04, p < 0.0001) in the ipsilateral hind paw of CCI mice (Fig. 1A and B; ##p < 0.01 vs VEH-treated CCI group on day 3).
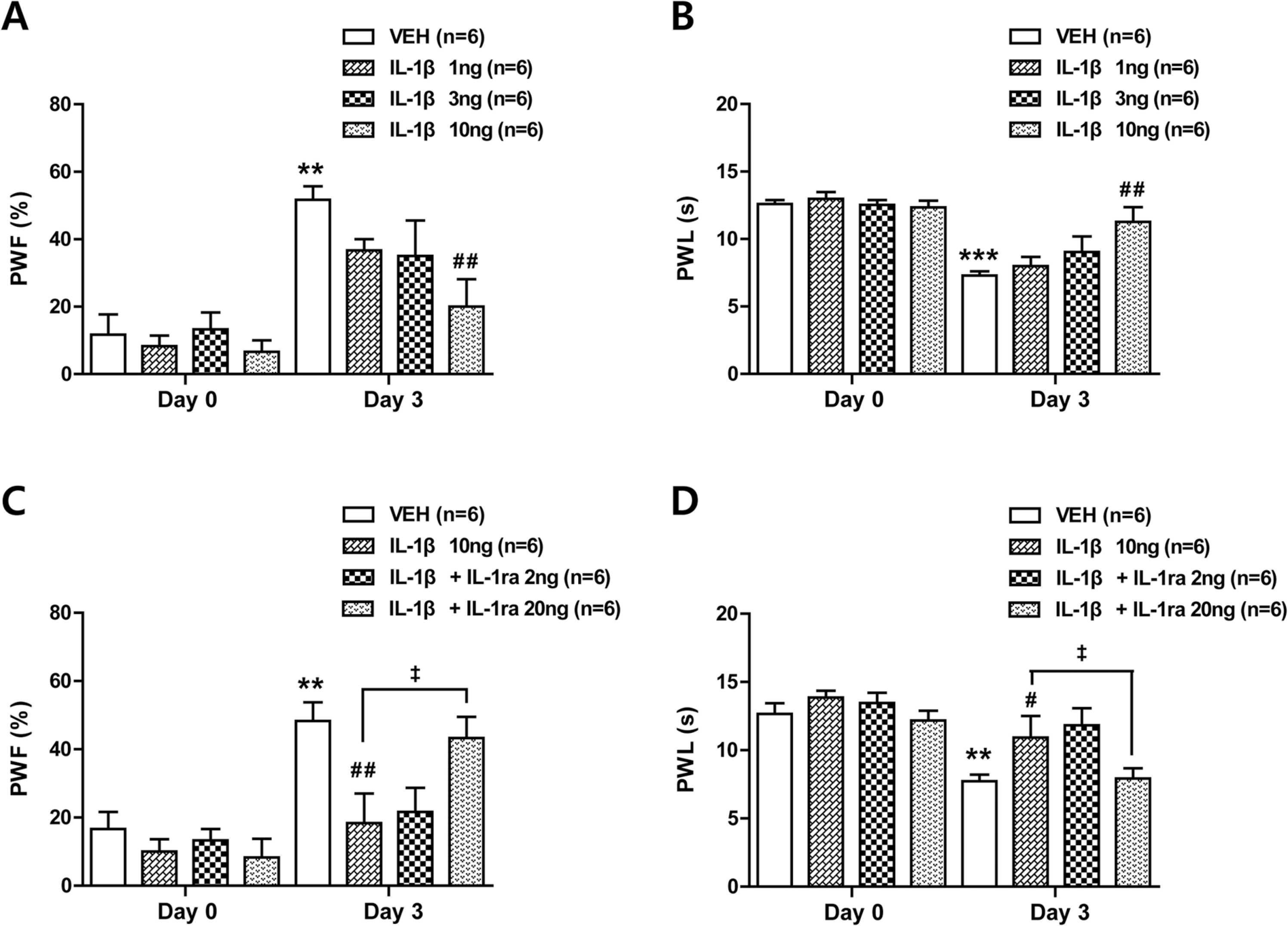
Effects of i.t. administration of IL-1β (1, 3 or 10 ng) in combination with an IL-1 receptor antagonist (IL-1ra, 2 or 20 ng) on the development of mechanical allodynia and thermal hyperalgesia in the ipsilateral hind paw of CCI mice. (A and B) Repeated daily administration of IL-1β dose-dependently inhibited the increase in paw withdrawal frequency (A; PWF, %) and the decrease in paw withdrawal latency (B; PWL, s) in the ipsilateral hind paw that occurred in VEH-treated CCI mice. (C and D) Administration of IL-1ra in combination with IL-1β restored the CCI-induced development of mechanical allodynia (C) and thermal hyperalgesia (D) in the ipsilateral hind paw. Drugs were administrated intrathecally from days 0–3 post-surgery. Mechanical allodynia and thermal hyperalgesia tests were performed at days 0 and 3 after nerve injury. n = 6 mice per experimental group. * * p < 0.01, * ** p < 0.001 vs VEH-treated CCI group on day 0; #p < 0.05, ##p < 0.01 vs VEH-treated CCI group on day 3; ‡p < 0.05 vs IL-1β-treated CCI group on day 3.
To verify whether the analgesic action of IL-1β is mediated by the direct activation of interleukin-1 receptors in the spinal cord, we administrated an interleukin-1 receptor antagonist (IL-1ra; 2 and 20 ng) with IL-1β during the induction phase of neuropathic pain. Co-administration of IL-1ra (20 ng) with IL-1β (10 ng) restored the development of mechanical allodynia (Fig. 1C; F (7,40) = 6.721, p < 0.0001) and thermal hyperalgesia (Fig. 1D; F (7,40) = 6.394, p < 0.0001) that was originally suppressed by IL-1β administration (Fig. 1C and D; ** p < 0.01 vs VEH-treated CCI group on day 0; #p < 0.05, ##p < 0.01 vs VEH-treated CCI group on day 3; ‡p < 0.05 vs IL-1β-treated CCI group on day 3). These results demonstrate that spinal IL-1β administration during the induction phase of neuropathic pain inhibits the development of neuropathic pain following CCI and that the actions of IL-1β are mediated by the direct activation of interleukin-1 receptors in the spinal cord.
3.2. I.t. administration of IL-1β reduces the CCI-induced increased pGluN1 expression in the ipsilateral spinal cord and this is restored by co-administration of IL-1ra with IL-1β
To determine whether spinal IL-1β has an effect on the functional potentiation of NMDA receptors in the spinal cord, we examined the changes in phosphorylation of NMDA receptor GluN1 subunit (pGluN1) using Western blot analysis. Sciatic nerve injury increased PKC-dependent pGluN1 expression at serine 896 site (Fig. 2A; * p < 0.05 vs Sham group; F (3,20) = 4.692, p = 0.0122) and PKA-dependent pGluN1 expression at serine 897 site (Fig. 2B; ** p < 0.01 vs Sham group; F (3,20) = 6.432, p = 0.0032) in the ipsilateral lumbar spinal cord dorsal horn. These increases were suppressed by repeated i.t. administration of IL-1β (10 ng) during the induction phase of neuropathic pain (Fig. 2A and B; #p < 0.05 vs VEH-treated CCI group). In addition, co-administration of IL-1ra (20 ng) with IL-1β (10 ng) restored the CCI-induced increase in PKC- and PKA-dependent pGluN1 expression that was originally suppressed by IL-1β administration (Fig. 2A and B; ‡p < 0.05 vs IL-1β-treated CCI group). These results demonstrate that spinal IL-1β administration during the early phase of neuropathic pain suppresses the CCI-induced increases in both PKC- and PKA-dependent phosphorylation of NMDA receptor GluN1 subunit.
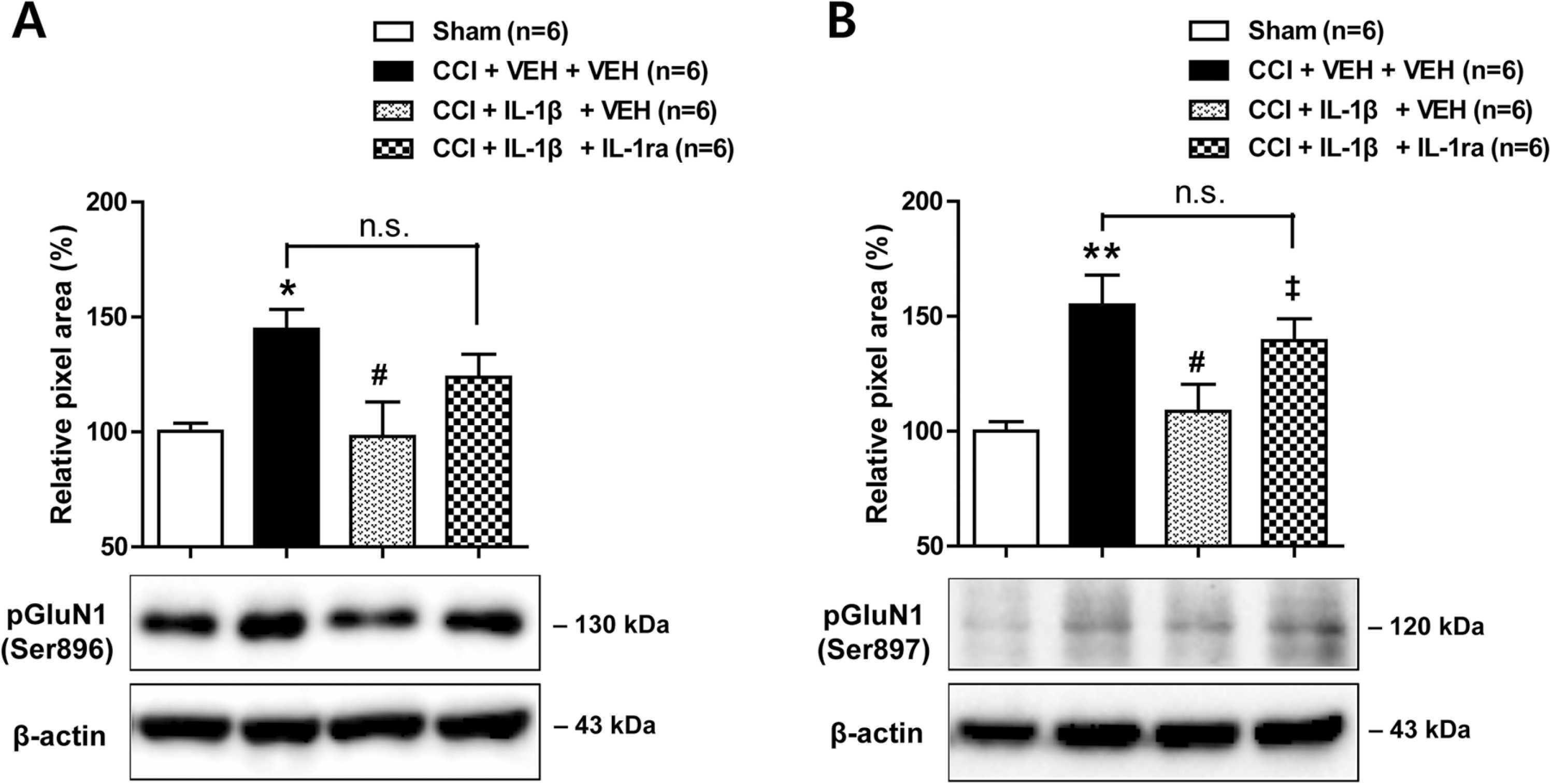
Effects of i.t. administration of IL-1β (10 ng) in combination with an IL-1 receptor antagonist (IL-1ra, 20 ng) on the expression of PKC- and PKA-dependent GluN1 phosphorylation (pGluN1) in the ipsilateral spinal cord dorsal horn of CCI mice. (A and B) Repeated daily administration of IL-1β (10 ng) reduced the CCI-induced increase in PKC-dependent pGluN1 expression at serine 896 site (A) and PKA-dependent pGluN1 expression at serine 897 site (B) in the ipsilateral spinal cord that occurred in VEH-treated CCI mice. Administration of IL-1ra in combination with IL-1β restored the CCI-induced increase in pGluN1 expression that was reduced by IL-1β. Drugs were administrated intrathecally from days 0–3 post-surgery. Lumbar spinal cord dorsal horn was sampled at day 3 after nerve injury. n = 6 mice per experimental group. * p < 0.05, * * p < 0.01 vs Sham group; #p < 0.05 vs VEH-treated CCI group; ‡p < 0.05 vs IL-1β-treated CCI group.
3.3. Neither CCI nor IL-1β administration has effects on pain sensitivity in the contralateral hind paw and pGluN1 expression in the contralateral spinal cord
By contrast, there was no significant change in the PWF to non-noxious mechanical stimulation (Fig. 3A; F (7,40) = 1.567, p = 0.1734) and the PWL to noxious thermal stimulation (Fig. 3B; F (7,40) = 0.7827, p = 0.6056) in the contralateral hind paw of CCI mice. In addition, there was no change in PKC-dependent pGluN1 expression at serine 896 site (Fig. 3C; F (3,20) = 0.2595, p = 0.8537) and PKA-dependent pGluN1 expression at serine 897 site (Fig. 3D; F (3,20) = 0.4489, p = 0.7208) in the contralateral lumbar spinal cord dorsal horn following CCI and drug administration.
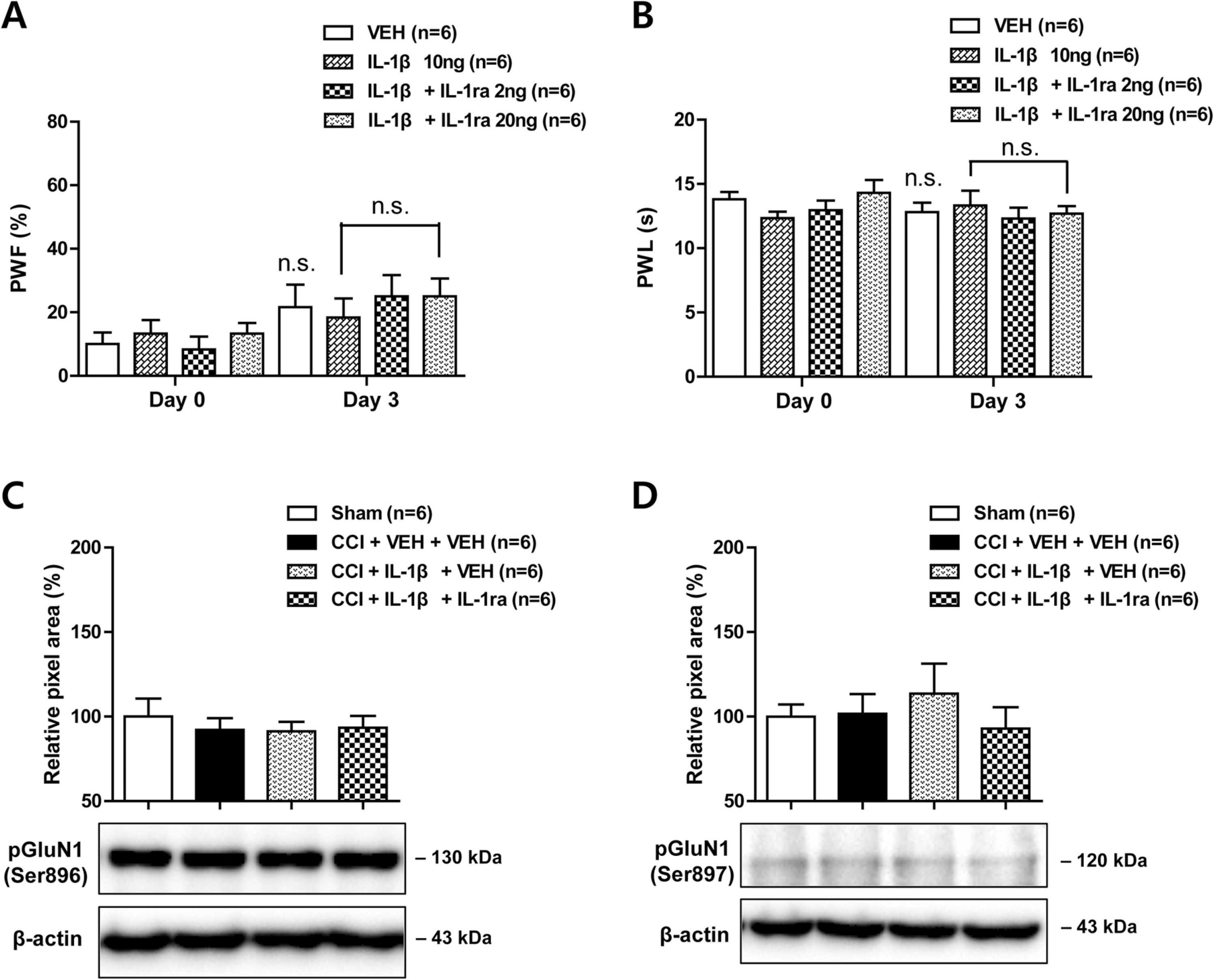
Effects of i.t. administration of IL-1β (10 ng) in combination with an IL-1 receptor antagonist (IL-1ra, 2 or 20 ng) on the development of mechanical allodynia and thermal hyperalgesia in the contralateral hind paw and on the expression of PKC- and PKA-dependent GluN1 phosphorylation (pGluN1) in the contralateral spinal cord dorsal horn of CCI mice. (A and B) The paw withdrawal frequency (A; PWF, %) and paw withdrawal latency (B; PWL, s) in the contralateral hind paw did not change following either CCI or drug administration. Mechanical allodynia and thermal hyperalgesia tests were performed at days 0 and 3 after nerve injury. (C and D) The PKC-dependent pGluN1 expression at serine 896 site (C) and PKA-dependent pGluN1 expression at serine 897 site (D) in the contralateral hind paw did not change in response to either CCI or drug administration. Drugs were administrated intrathecally from days 0–3 post-surgery. Lumbar spinal cord dorsal horn was sampled at day 3 after nerve injury. n = 6 mice per experimental group. n.s., not significant.
3.4. Sciatic nerve injury-induced increase in astrocyte sigma-1 receptor expression is inhibited by i.t. administration of IL-1β
We have previously demonstrated that the spinal Sig-1R plays an important role in NMDA receptor phosphorylation [24], [25]. Thus, we next examined whether spinal IL-1β changes the expression of Sig-1R in the lumbar spinal cord dorsal horn of CCI mice using Western blot analysis and immunohistochemistry. Sciatic nerve injury increased the expression of Sig-1R in the ipsilateral spinal cord dorsal horn at day 3 post-surgery and this increase was significantly suppressed by repeated administration of IL-1β (10 ng) from days 0–3 post-surgery (Fig. 4A; * p < 0.05 vs Sham; #p < 0.05 vs VEH-treated CCI group; F (2,15) = 5.630, p = 0.0150). By contrast, sciatic nerve injury and IL-1β administration had no effect on the expression of Sig-1R in the contralateral spinal cord dorsal horn at day 3 post-surgery (Fig. 4B; F (2,15) = 0.1994, p = 0.8213).
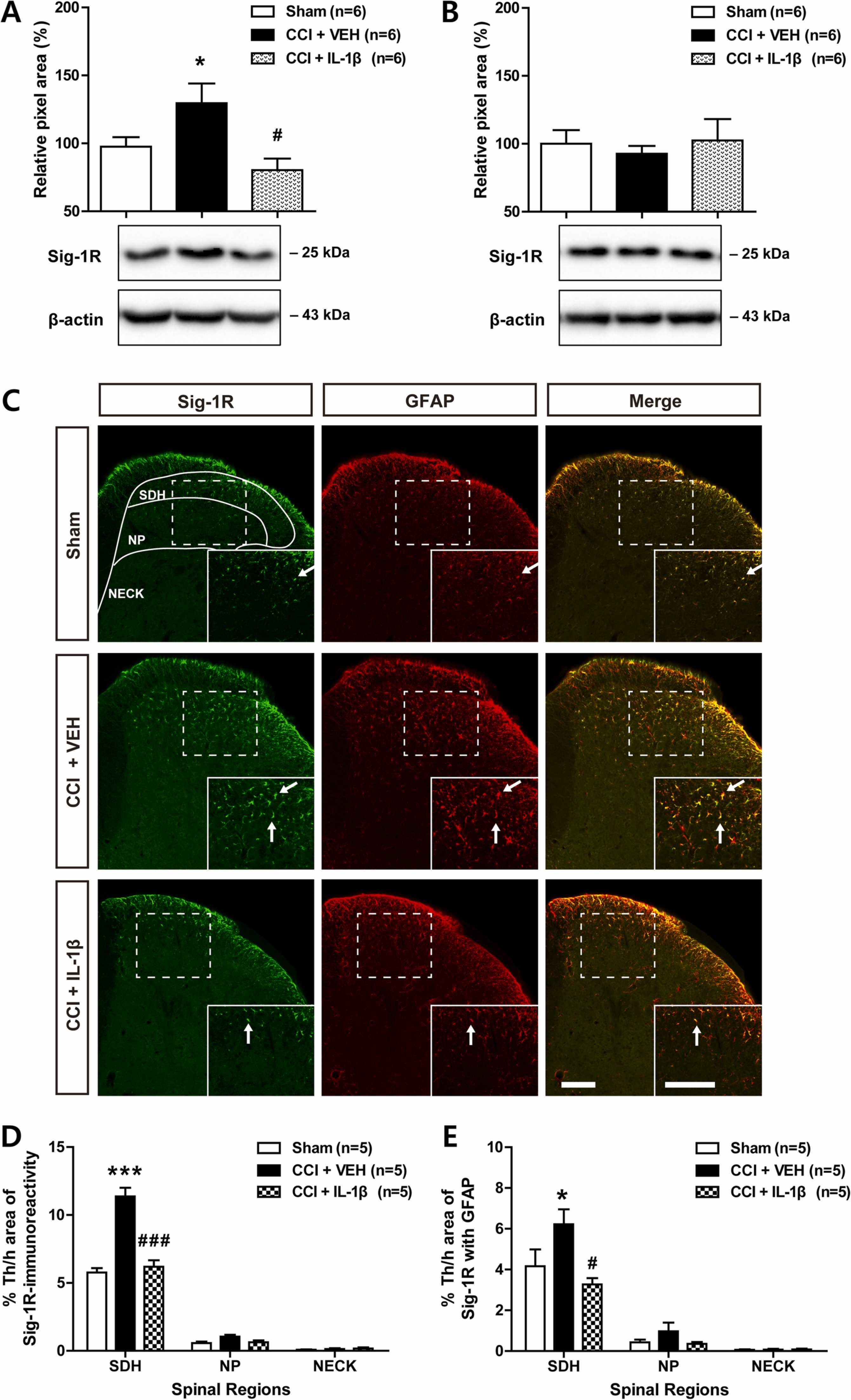
Effects of i.t. administration of IL-1β (10 ng) on the expression of Sig-1R in the spinal cord dorsal horn of CCI mice. (A and B) Western blot analysis showed that the sciatic nerve injury-induced increase in Sig-1R expression in the ipsilateral dorsal horn (A) was suppressed by IL-1β administration, while the expression of Sig-1R in the contralateral dorsal horn (B) did not change following CCI and/or IL-1β administration. n = 6 mice per experimental group. (C–E) Lumbar spinal cord sections were immunostained for Sig-1R (green) and GFAP. Representative confocal images are shown in C. The ipsilateral spinal cord dorsal horn was analyzed in the superficial dorsal horn (SDH, laminae I-II), nucleus proprius (NP, laminae III-IV), and neck region (NECK, laminae V-VI). Arrows indicate colocalization of Sig-1R with GFAP immunostaining. Scale bar = 100 µm. Repeated daily administration of IL-1β reduced the increases in Sig-1R immunoreactivity (D, green) and Sig-1R immunoreactivity in GFAP-positive astrocytes (E, yellow) that occurred in VEH-treated CCI mice. Drugs were administrated intrathecally from days 0–3 post-surgery. The lumbar spinal cord dorsal horn was sampled at day 3 after nerve injury. n = 5 mice per experimental group. * p < 0.05, * ** p < 0.001 vs Sham group; #p < 0.05, ###p < 0.001 vs VEH-treated CCI group.
Representative ipsilateral lumbar spinal cord dorsal horn sections are shown in the Fig. 4C. Sciatic nerve injury increased the expression of Sig-1R immunoreactivity in the SDH region of the ipsilateral lumbar spinal cord and this increase was significantly reduced by repeated i.t. administration of IL-1β (10 ng) during the induction phase of neuropathic pain (Fig. 4D; *** p < 0.001 vs Sham group; ###p < 0.001 vs VEH-treated CCI group; SDH: F (2,12) = 39.10, p < 0.0001, NP: F (2,12) = 3.994, p = 0.0468, NECK: F (2,12) = 0.3856, p = 0.6882). In addition, sciatic nerve injury increased Sig-1R immunoreactivity in GFAP-positive astrocytes located in the SDH region and this increase was suppressed by i.t. administration of IL-1β (10 ng) during the induction phase of neuropathic pain (Fig. 4E; * p < 0.05 vs Sham group; #p < 0.05 vs VEH-treated CCI group; SDH: F (2,12) = 5.238, p < 0.0232, NP: F (2,12) = 1.611, p = 0.2400, NECK: F (2,12) = 0.06770, p = 0.9349). These results show that spinal IL-1β administration influences the expression of Sig-1R, which is increased in astrocytes located in SDH region of the ipsilateral spinal cord in CCI mice.
3.5. Co-administration of PRE084 with IL-1β restores the development of mechanical allodynia and spinal pGluN1 expression which are suppressed by IL-1β administration in CCI mice
In order to verify whether agonistic stimulation of the Sig-1R restores the analgesic effect of IL-1β on the development of neuropathic pain, the Sig-1R agonist, PRE084 (1 or 10 nmol) was co-administrated intrathecally with IL-1β during the induction phase of neuropathic pain in CCI mice. Co-administration of PRE084 (10 nmol) with IL-1β (10 ng) restored the development of mechanical allodynia in the ipsilateral hind paw, which was originally suppressed by IL-1β administration (Fig. 5A; *** p < 0.001 vs VEH-treated CCI group on day 0; ##p < 0.01 vs VEH-treated CCI group on day 3; ‡p < 0.05 vs IL-1β-treated CCI group on day 3; F (7,40) = 14.15, p < 0.0001). By contrast, the suppressive effect of IL-1β on the development of thermal hyperalgesia did not change by co-administration of PRE084 with IL-1β (Fig. 5B; * p < 0.05 vs VEH-treated CCI group on day 0; #p < 0.05 vs VEH-treated CCI group on day 3; F (7,40) = 3.404, p = 0.0060).
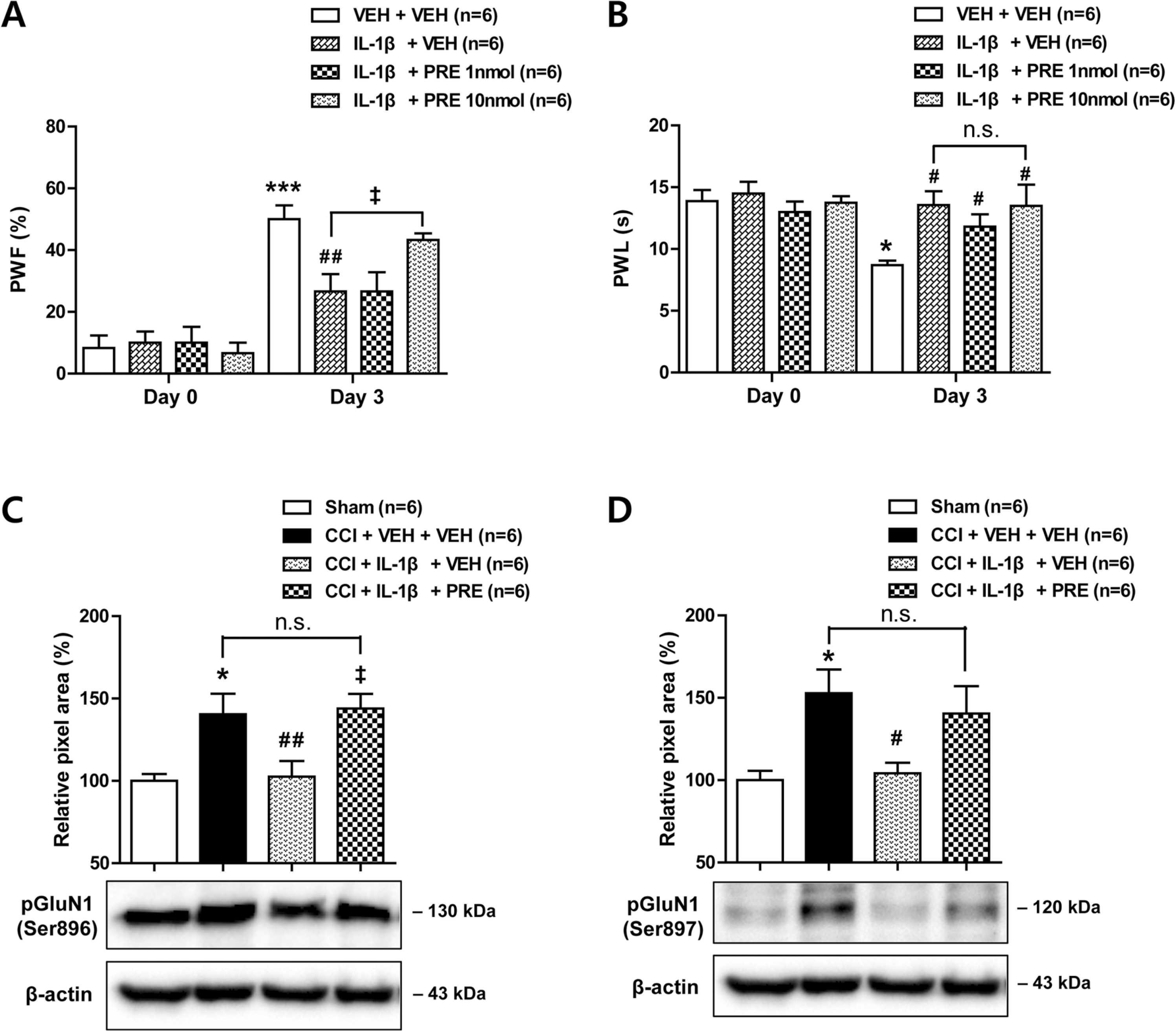
Effect of i.t. administration of the Sig-1R agonist, PRE084 (PRE; 1 or 10 nmol) in combination with IL-1β (10 ng) on the development of neuropathic pain and on the expression of PKC- and PKA-dependent GluN1 phosphorylation (pGluN1) in CCI mice. (A and B) Repeated daily administration of PRE084 (10 nmol) in combination with IL-1β restored the CCI-induced increase in paw withdrawal frequency (A; PWF, %) and the decrease in paw withdrawal latency (B; PWL, s) in the ipsilateral hind paw that were inhibited by IL-1β administration. * p < 0.05, * ** p < 0.001 vs VEH-treated CCI group on day 0; #p < 0.05, ##p < 0.01 vs VEH-treated CCI group on day 3; ‡p < 0.05 vs IL-1β-treated CCI group on day 3. (C and D) Western blot analysis showed that administration of PRE084 (10 nmol) in combination with IL-1β (10 ng) restored the CCI-induced increase in PKC-dependent (C) and PKA-dependent (D) phosphorylation of the GluN1 subunit in the ipsilateral spinal cord dorsal horn. The lumbar spinal cord dorsal horn was sampled at day 3 after nerve injury. * p < 0.05 vs Sham group; #p < 0.05, ##p < 0.01 vs VEH-treated CCI group; ‡p < 0.05 vs IL-1β-treated CCI group. Drugs were administrated intrathecally from days 0–3 post-surgery. n = 6 mice per experimental group.
Since spinal Sig-1Rs play an important role in the CCI-induced increases in PKC- and PKA-dependent phosphorylation of the NMDA receptor GluN1 subunit, we next examined whether spinal Sig-1R stimulation also restores the inhibitory effect of IL-1β on the CCI-induced increase in pGluN1 expression. Co-administration of PRE084 (10 nmol) with IL-1β (10 ng) restored both PKC-dependent pGluN1 expression at the serine 896 site (Fig. 5C; * p < 0.05 vs Sham group; ##p < 0.01 vs VEH-treated CCI group; ‡p < 0.05 vs IL-1β-treated CCI group; F (3,20) = 6.495, p = 0.0030) and PKA-dependent pGluN1 expression at the serine 897 site (Fig. 5D; * p < 0.05 vs Sham group; #p < 0.05 vs VEH-treated CCI group; F (3,20) = 4.964, p = 0.0098) in the ipsilateral spinal cord dorsal horn, which were originally suppressed by IL-1β administration alone. These results demonstrate that agonistic stimulation of spinal Sig-1Rs restores the inhibitory effect of IL-1β on the development of mechanical allodynia and GluN1 phosphorylation in the ipsilateral spinal cord following peripheral nerve injury.
4. Discussion
There are three important novel findings in the present study that support our original hypothesis. First, repeated daily i.t. administration of IL-1β from days 0–3 post-surgery (during the induction phase of neuropathic pain) significantly inhibited the CCI-induced increased phosphorylation of the NMDA receptor GluN1 subunit (pGluN1) in the ipsilateral spinal cord, as well as, the development of neuropathic pain in the ipsilateral hind paw. Moreover, this inhibitory effect of IL-1β was restored by blockade of the IL-1 receptor with IL-1ra administration. Secondly, sciatic nerve injury increased the expression of the Sig-1R in ipsilateral spinal cord astrocytes at day 3 post-surgery, and this increase was reduced by repeated administration of IL-1β. Finally, agonistic stimulation of Sig-1Rs with PRE084 from days 0–3 post-surgery restored pGluN1 expression and mechanical allodynia development that were originally suppressed by i.t. administration of IL-1β alone. Collectively these results demonstrate that administration of exogenous IL-1β during the early phase of neuropathic pain has an inhibitory effect on phosphorylation of the GluN1 subunit, as well as the development of mechanical allodynia, and these actions of IL-1β are mediated via regulation of spinal astrocyte Sig-1R expression following peripheral neuropathy.
Interleukin-1 binds to two types of receptors on the cell membrane; IL-1 receptor type 1 (IL-1R1) and IL-1 receptor type 2 (IL-1R2) [26]. IL-1R1 is responsible for IL-1 signal transduction, whereas IL-1R2 is a decoy receptor that serves as an endogenous inhibitor of IL-1 signaling [27]. In a previous study we showed that IL-1R1 is mainly co-localized with GFAP-positive astrocytes in the SDH region of the spinal cord suggesting the close interaction of IL-1 signaling with spinal astrocytes located in superficial dorsal horn following nerve injury or inflammation [8], [9]. IL-1α or IL-1β is known to serve as activators of the IL-1R1, while the IL-1 receptor antagonist (IL-1ra) is a competitive inhibitor that prevents IL-1-dependent intracellular signaling [27]. In the present study, IL-1β-induced modulation of the development of neuropathic pain was restored by blockade of the interleukin-1 receptor with i.t. administration of IL-1ra. Thus, it could be possible that injection of IL-1β during the early phase of neuropathic pain stimulates interleukin-1 receptors in the SDH region of the spinal cord and that this IL-1 receptor-mediated signaling plays an important role in both the control of pathological changes in astrocytes and nerve injury-induced pain signaling transmission.
Central sensitization, refers to an increased responsiveness of spinal cord dorsal horn neurons to nociceptive peripheral stimulation and this phenomenon has been strongly implicated in the generation of chronic pain [28]. The activation of NMDAreceptors is critically involved with both the induction and maintenance of central sensitization [29]. It has been suggested that PKC-dependent phosphorylation increases the opening rate of NMDA channels and the delivery of functional NMDA receptors to the cell surface [30], [31]. Moreover, PKA activation has been shown to increase the amplitude of NMDA receptor-mediated excitatory postsynaptic currents, synaptic targeting of NMDA receptors, and Ca2+ permeability through NMDA channels [32], [33], [34] resulting in increase of synaptic strength. It has also been shown that the Sig-1R enhances NMDA receptor synaptic transmission and plasticity in rat hippocampus [35]. In addition, our previous results indicated that activation of spinal Sig-1Rs increase serine racemase expression in astrocytes and concomitant D-serine production, which in turn mediates Sig-1R-induced functional potentiation of NMDA receptors via an increase in spinal NMDA receptor phosphorylation [25], [36]. In the present study, IL-1β administration inhibited the CCI-induced increase in astrocyte Sig-1R expression, and agonistic stimulation of the Sig-1R via administration of the Sig-1R agonist, PRE084 restored the CCI-induced development of mechanical allodynia and NMDA receptor phosphorylation that were significantly decreased by IL-1β administration alone. These results suggest that the IL-1β-induced inhibition of astrocyte Sig-1R expression may play a critical role in regulating the potentiation of NMDA receptors-mediated synaptic transmission and the development of mechanical allodynia in CCI mice.
In the present study we focused on the inhibitory effect of spinal IL-1β on the development of neuropathic pain during the early phase of peripheral nerve injury. Our results are consistent with the data from Souter and colleagues suggesting that i.t. IL-1β suppresses carrageenan-induced inflammatory pain by a non-opioid mechanism at the level of the spinal cord [4]. It has been also suggested that intraplantar administration of recombinant human IL-1β activates its receptor on immune cells to release opioids that bind multiple opioid receptors on sensory nerves resulting in antinociception in a rat model of Freund’s complete adjuvant-induced inflammatory pain [37]. By contrast, when administered into a variety of peripheral tissues, IL-1β has been shown to be nociceptive. IL-1β decreased voltage-gated K+ currents in the small-diameter trigeminal ganglion neurons and potentiated neuronal excitability leading to trigeminal inflammatory hyperalgesia[6]. IL-1β increased the excitability of adult dorsal root ganglion nociceptor-like neurons in vitro and intraplantar administration of IL-1β produced mechanical and thermal pain hypersensitivity [7]. In addition, it has been reported that IL-1β increased the frequency and amplitude of spontaneous excitatory postsynaptic currents (EPSCs) as well as NMDA-induced currents in lamina II neurons of isolated spinal cord slices from naïve rats [38]. This discrepancy may be explained by one or more of the following differences: 1) animal models of injury used, 2) the time course of ongoing pain, 3) doses, routes, and the timing of the treatment, and 4) examined nervous system locations. These differences may affect not only the interaction between this cytokine and its receptor located on glial cells and neurons, but also the intracellular response depending on the cell type affected. Since the effect of IL-1β can be nociceptive or antinociceptive, the detailed mechanisms need to be further explored.
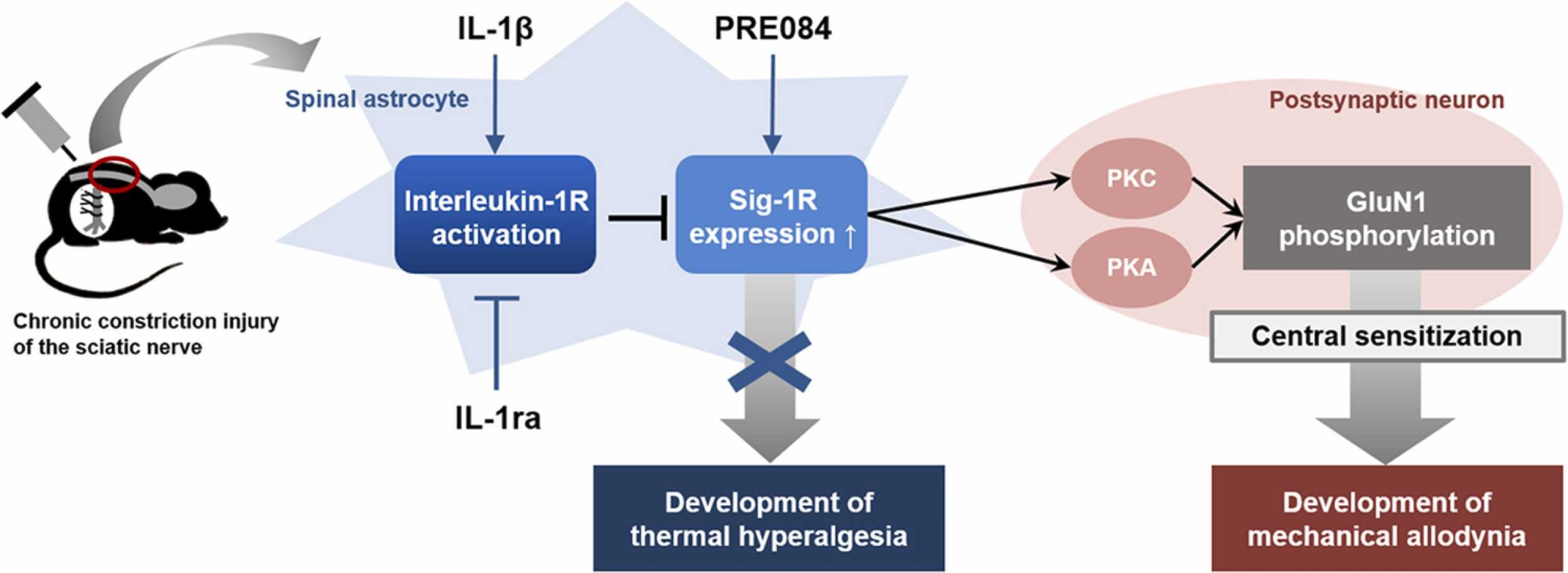
In conclusion, the present study demonstrates that intrathecal administration of IL-1β during the induction phase of neuropathic pain significantly reduces the CCI-induced development of neuropathic pain, as well as, the increase in NMDA receptor GluN1 phosphorylation in the spinal cord. Furthermore, these actions of IL-1β are mediated through the control of the spinal astrocyte Sig-1R expression (Fig. 6). These results support our hypotheses and raise the possibility that administration of exogenous IL-1β during the early phase of neuropathic pain could be a novel therapeutic approach to control the development of neuropathic pain associated with peripheral neuropathy.
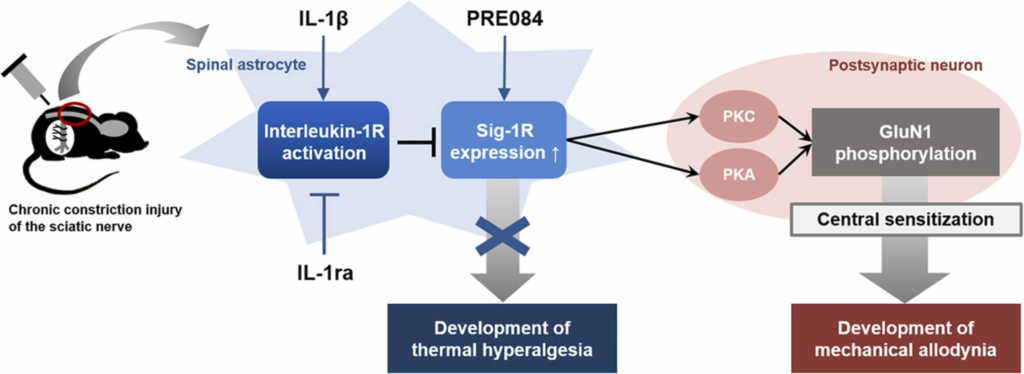
Schematic diagram summarizing the proposed mechanisms underlying interleukin-1β (IL-1β)-induced inhibition of the development of neuropathic pain in CCI mice. Stimulation of spinal interleukin-1 receptors with intrathecal administration of IL-1β decreases the expression of Sig-1Rs that are increased in spinal astrocytes following peripheral nerve injury. This chage leads to a decrease in PKC- and PKA-dependent phosphorylation of the NMDA receptor GluN1 subunit in postsynaptic neurons via indirect methods, and ultimately controls the development of mechanical allodynia in CCI mice. By contrast, IL-1β-induced inhibition of the development of thermal hyperalgesia is not likely to be mediated by Sig-1R activation.