
Interorganelle communication, aging, and neurodegeneration

By Maja Petkovic, Caitlin E. O’Brien, and Yuh Nung Jan
Excerpt from the article published in Genes & Dev. 2021. 35: 449-469 PMCID: PMC8015714 DOI: 10.1101/gad.346759.120
Editor’s Highlights
- Cellular organelles and their associated processes do not function in isolation, but within a robust cellular communication network—the cellular communicome—to maintain homeostasis.
- Many proteins participate at multiple distinct contact sites between different pairs of organelles.
- Perturbation of Membrane contact sites, in particular those between the ER and mitochondria called MERCs, are a major driver of aging and various age-related pathologies.
- The σ1 receptor (Sig1R), an ER chaperone with cytoprotective activity, localizes to MERCs and can stabilize IP3R.
- Mutations in the SIGMAR1 gene encoding Sig1R are Directly Linked with Aging.
Abstract
Our cells are comprised of billions of proteins, lipids, and other small molecules packed into their respective subcellular organelles, with the daunting task of maintaining cellular homeostasis over a lifetime. However, it is becoming increasingly evident that organelles do not act as autonomous discrete units but rather as interconnected hubs that engage in extensive communication through membrane contacts. In the last few years, our understanding of how these contacts coordinate organelle function has redefined our view of the cell. This review aims to present novel findings on the cellular interorganelle communication network and how its dysfunction may contribute to aging and neurodegeneration. The consequences of disturbed interorganellar communication are intimately linked with age-related pathologies. Given that both aging and neurodegenerative diseases are characterized by the concomitant failure of multiple cellular pathways, coordination of organelle communication and function could represent an emerging regulatory mechanism critical for long-term cellular homeostasis. We anticipate that defining the relationships between interorganelle communication, aging, and neurodegeneration will open new avenues for therapeutics.
Interorganelle contacts, a historical perspective
Eukaryotic cells are defined by the presence of multiple intracellular organelles, each specialized for a unique function, that collectively coordinate the cell’s response to external and internal changes to sustain life. To do so, an organelle communicates with its environment by importing and exporting material through membrane transporters, as well as by exchanging material with other organelles through vesicular transport. However, mitochondria, peroxisomes, and plastids are not connected to major vesicular transport systems (Sugiura et al. 2014). Furthermore, vesicular transport cannot account for the exchange of certain biomolecules. For example, delivery of several classes of lipids from their site of synthesis in the endoplasmic reticulum (ER) to their steady-state location is unaffected by pharmacological and genetic manipulations that deplete cellular ATP levels, disrupt vesicular transport, and alter cytoskeletal dynamics (Kaplan and Simoni 1985; Vance et al. 1991; Hanada et al. 2003; Baumann et al. 2005; Lev 2012). Moreover, intracellular compartments like the ER and mitochondria refill their depleted calcium (Ca2+) stores through low-affinity calcium channels that require much higher local concentrations of calcium than global cytosolic Ca2+ levels (Jousset et al. 2007; Carrasco and Meyer 2011). These findings raise the question about the existence of other communication pathways such as direct contacts between organelles, and between intracellular compartments and the plasma membrane.
Such “intimate associations” were observed already in the 1950s in meticulous electron microscopy studies (Porter and Palade 1957; Rosenbluth 1962). However, without an established physiological relevance and known molecular components, such observations were mainly disregarded as artifacts, a consequence of a dense cellular environment or a feature only of highly specialized cells. It was not until a seminal paper by Jean Vance in 1990 that the first function of contact sites was reported. Using purified subcellular fractions of ER mitochondria-associated membranes, named MAM, she established these organelle contacts between the ER and mitochondria as sites of lipid synthesis and transfer (Vance 1990). A few years later, MAMs were implicated as sites of Ca2+homeostasis (Rizzuto et al. 1998). It became apparent that organelles often form such contact sites, defined as specialized microdomains of close apposition within the range of protein–protein interactions that allow for coordination of organellar functions without organelle fusion. The identification of proteins mediating such interorganelle contacts was the next leap for the field, with those mediating ER-mitochondria contacts identified first. The mitofusins were found to form contacts required for Ca2+ homeostasis (de Brito and Scorrano 2008), while the multisubunit ERMES (ER-mitochondria encounter structure) complex is required for phospholipid biosynthesis (Kornmann et al. 2009). Today, the steady discovery of novel interoganelle contacts, the proteins that mediate them, and their function has brought this field from obscurity to center stage. An exhaustive description of all contact sites and the proteins that regulate them is well beyond the scope of this review, and the interested reader is referred to many excellent reviews that provide detailed analyses of this topic (Eisenberg-Bord et al. 2016; Scorrano et al. 2019). Our apologies to colleagues whose work we did not highlight in this review.
The redefined view of the cell as interconnected network
The current view of the cell is that interorganelle communication through contact sites is ubiquitous across all eukaryotic organisms, cell types, and organelles (Gatta and Levine 2017). While originally called membrane contact sites, monobilayer organelles like lipid droplets, as well as nonmembranous organelles like RNA granules (Liao et al. 2019) or stress granules (Lee et al. 2020), also engage in interorganelle communication. It has been proposed that contact sites predate vesicular trafficking, emerging early in eukaryotic cells where they could have played a crucial role in the acquisition of bacteria-type lipids from protomitochondria by their archaeal host (Jain and Holthuis 2017). As eukaryotes evolved to contain the full suite of organelles we recognize today, so did the contacts between them also evolve to facilitate an increasing number of functions. For example, organelles exert physical forces on each other (Helle et al. 2017; Feng and Kornmann 2018), and in the dense intracellular environment, contact sites may have evolved to harness those forces for organelle remodeling, determining the sites of fission for endosomes and mitochondria (Friedman et al. 2011; Rowland et al. 2014; Wong et al. 2018). In such a dense eukaryotic cell, interorganelle communication is spatially and temporally organized. Organelles have a characteristic distribution and dispersion pattern in three-dimensional space, and there is a reproducible pattern of transient dynamic contacts between distinct organelles that responds to cell physiological conditions (Friedman et al. 2011; Rowland et al. 2014; Wu et al. 2017; Wong et al. 2018). For example, contact sites between ER and lysosomes respond to cholesterol levels, enabling activation of master growth regulator mTORC1 kinase (Lim et al. 2019), while ER-mitochondria contacts form in response to hypoxia (Wu et al. 2016). Different organelle pairs also have different frequencies of contacts. It is important to mention that interorganelle communication at contact sites is just one of several systems underlying the functional interdependence of organelles. Material exchange through contact sites is spatially segregated (Wu et al. 2017; Ng et al. 2018; Chung et al. 2020) but mutually coordinated (Petkovic et al. 2014; Gallo et al. 2019; Weber-Boyvat et al. 2020) with vesicular transport. These systems also integrate signaling and transcriptional programs to support cellular homeostasis and survival.
Aging and age-related pathology as a consequence of disturbed interorganelle communication
Communication between organelles allows them to function and adapt to changing cellular environments to maintain cellular homeostasis. The gradual failure of the cell’s ability to maintain homeostasis leads to physiological aging and age-related pathologies like neurodegenerative diseases. Decades of research have identified the mechanisms underlying physiological aging with relevance to pathological aging, which include dysregulated nutrient sensing, dysfunctional bioenergetics, defective protein quality control and degradation, the seeding and propagation of stress granules, and maladaptive innate immune responses (López-Otín et al. 2013; Gan et al. 2018). Only more recently has interorganelle communication been implicated in many of these processes: proper functioning of lipid metabolism, mitochondrial function, and endolysosomal function. Here we highlight novel findings that link defects of interorganelle communication to aging and neurodegeneration. While the loss of very few genes involved in contact sites is developmentally lethal, several genes associated with age-related pathologies have been directly linked to the failure of distinct contact sites, as we elaborate on in “Contact Sites Directly Linked with Aging and Neurodegeneration,” below (Fig. 1). Likewise, the morphology and physiology of contact sites is perturbed in a variety of age-related neurodegenerative diseases, as we discuss in “Contribution of Dysfunctional Interorganelle Communication to Aging and Neurodegeneration,” below (Fig. 6, below). While there is still more work needed to disentangle whether these contact site disturbances are a cause or consequence of disease, these correlative observations suggest that coordination of organelle communication at sites of contact could be an emerging regulatory mechanism for long-term cellular homeostasis. Because contact sites allow for direct communication between organelles, they could play a central role in a cell’s ability to respond to and compensate for functional changes in a given organelle or physiological process. Breakdown of such communication between organelles could progressively and synergistically drive cellular deterioration in aging and age-related processes. We accentuate the need to further understand these contacts, how they relate to one another and to the functioning of organelles and the cell as a whole. Last, we discuss how the conceptual shift from reductionism to a systems approach could provide insights into how collapse of interorganelle communication may contribute to aging and neurodegeneration, and reveal potential relevant therapeutic targets.
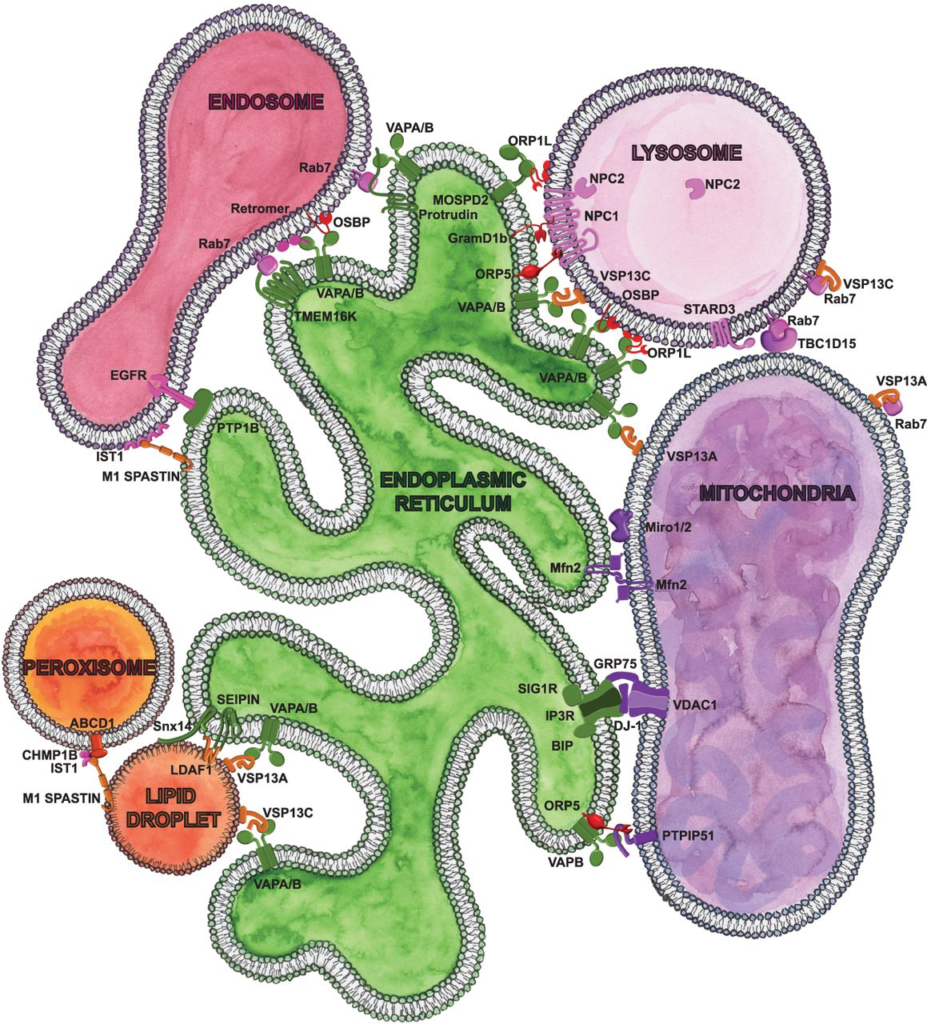
Scheme of cellular communicome highlighting the contact sites and proteins implicated in age and age-related pathologies presented in this review. Please note that we presented only a subset of contacts and contact site proteins. ER-localized proteins are labeled in green. Lysosome proteins are labeled in pink. Cytosolic lipid transfer proteins are labeled in red. Mitochondria proteins are labeled in purple. Lipid droplet localized proteins are labeled in orange. Peroxisome proteins are red, and endosome proteins are fushia. Proteins can be localized to multiple organelles and participate at multiple distinct contact sites.
Contact sites directly linked with aging and neurodegeneration
Contact sites involved in lipid metabolism
Lipid metabolism has an important role in aging and neurodegeneration, with multiple cellular organelles coordinating key functions. Out of all the lipid species, cholesterol homeostasis is indubitably the most well-studied (Anchisi et al. 2012; Johnson and Stolzing 2019). Cholesterol is essential for the organization of cell membranes and serves as a precursor of several classes of signaling molecules. It is acquired both from endogenous synthesis and from diet. Dietary low-density lipoprotein (LDL)-derived cholesterol is endocytosed and transferred to the ER, which senses cholesterol levels and down-regulates endogenous synthesis in response. Under physiological conditions of dietary sterol depletion, cholesterol is transported in the opposite direction, from the site of endogenous synthesis in the ER to endolysosomal compartments. Contact sites between the ER and endolysosomal compartments have emerged as essential for bidirectional nonvesicular transfer of cholesterol (Fig. 2). NPC1, a major cholesterol transporter localized to the limiting membrane of late endosome/lysosomes, mediates contacts with the ER through interaction with the lipid transfer proteins ORP5 (Du et al. 2011) or Gramd1b (Höglinger et al. 2019). In coordination with NPC2, NPC1 transfers cholesterol from the lysosomal lumen to the cytosolic leaflet (Winkler et al. 2019), making cholesterol available for nonvesicular transport to the ER by lipid transfer proteins (Infante et al. 2008). Lipid transfer proteins, like ORP5 and Gramd1b, then bind monomeric lipids in a hydrophobic pocket and transfer them between the two membranes through an aqueous phase at the membrane contact site.
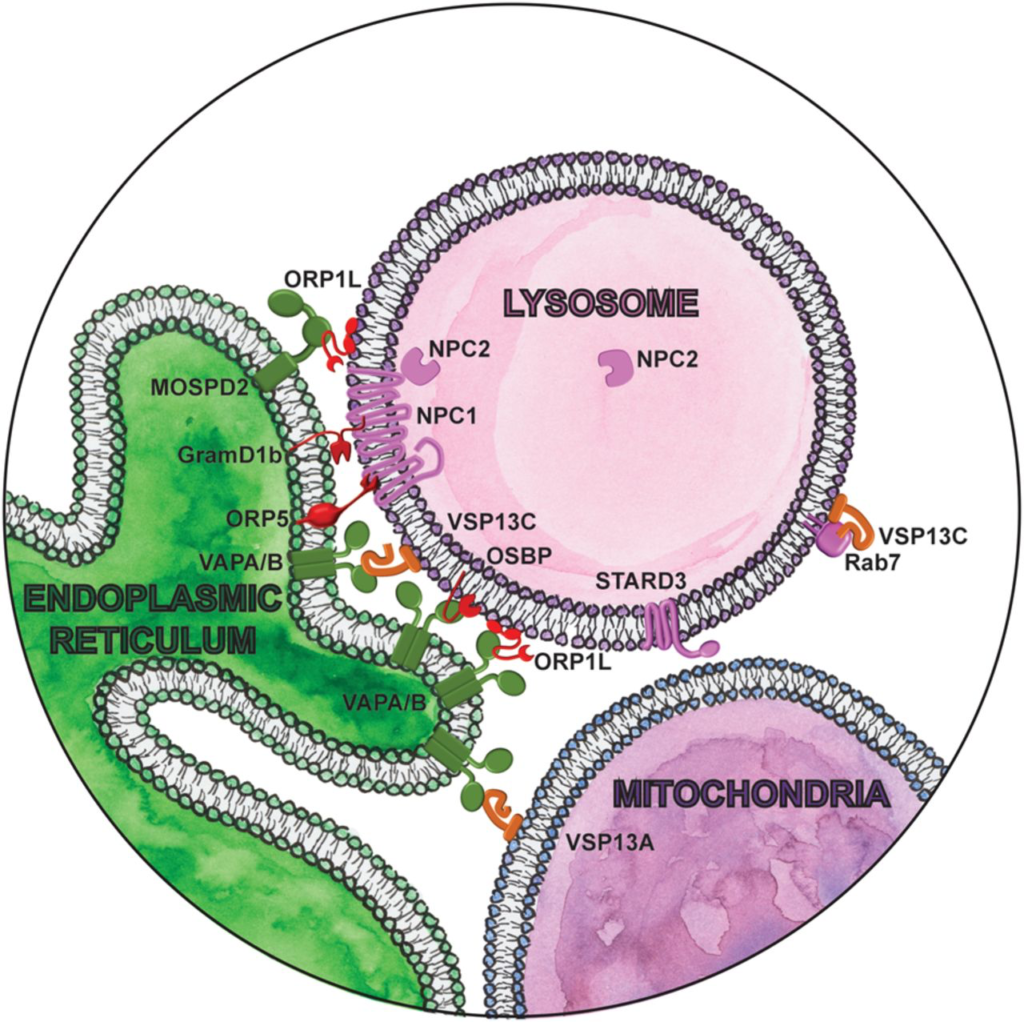
Magnifying glass view of the contact sites between the ER and lysosomes. ER-localized proteins are labeled in green. Lysosome proteins are labeled in pink. Cytosolic lipid transfer proteins are labeled in red. Lipid droplet localized proteins are labeled in orange.
Mutations in NPC1 and NPC2 cause Niemann-Pick disease type C, a lysosomal storage disorder that causes progressive neurodegeneration (Sturley et al. 2004). In cells lacking NPC1, cholesterol egress from the lumen of lysosomes to the ER is blocked. However, the effect of NPC1 deletion or mutation on the number and persistence of ER-lysosome contacts varies between studies, likely reflecting the complexity and dynamic nature of these contacts, as well as our technical limitations in studying them (Enrich et al. 2019). What is clear is that other distinct contact sites can aggravate or compensate for the loss of NPC1. OSBP1-mediated cholesterol transfer from the ER to endolysosomes is increased in NPC1-null cells, resulting in aberrant mTORC1 activation (Lim et al. 2019). Pharmacological suppression of mTORC1 signaling can correct several aspects of cellular function independent of the cholesterol storage defect, thus implicating dysregulated mTORC1 signaling as a likely driver of pathology in Niemann-Pick type C (Davis et al. 2021). Similarly, another lipid transfer protein ORP1L mediates bidirectional cholesterol transfer between the ER and endolysosomes through interaction with three different ER receptors: VAPA, VAPB, or MOSPD2 (Rocha et al. 2009; Zhao and Ridgway 2017; Di Mattia et al. 2018; Zhao et al. 2020). Forcing ER-endolysosomal contacts by overexpressing ORP1L can rescue lysosomal cholesterol levels in NPC1-null cells (Höglinger et al. 2019). Likewise, cells respond to lysosomal cholesterol accumulation by increasing lysosome-mitochondrial contacts dependent on sterol transfer protein STARD3 (MLN64) (Höglinger et al. 2019). Sterol accumulation in mitochondria appears to be a distinguishing feature of Niemann-Pick type C (Charman et al. 2010), as it is not observed in type A or B patients with mutations in the lysosomal enzymes responsible for lipolysis (Torres et al. 2017). This suggests that it may be a specific response to disruption of ER-endolysosome contacts and not simply the aberrant build-up of endolysosomal cholesterol and sphingolipids. Whether ER-endolysosome contacts increase in Niemann-Pick type A and B patients to facilitate egress of excess cholesterol from lysosomes is unknown. Cholesterol transport between other organelles is mediated by other distinct contact sites (Eden et al. 2016; Wilhelm et al. 2017), and it remains to be determined how they are all coordinated in maintaining cholesterol homeostasis.
Excess fatty acids and cholesterol are stored in another organelle, lipid droplets (LDs), in their esterified forms as neutral lipids, triacylglycerols (TAGs), and sterol esters, respectively. LDs serve as an energy reservoir for the cell during changes in cell growth and energy demand (Farese and Walther 2009). Contacts between LDs and the ER are essential for lipid storage as they enable de novo LD biogenesis and maturation (Fig. 3). Sites of such LD biogenesis are determined by locally concentrating TAGs leading to formation of contact sites between oligomers of the integral ER protein seipin and newly LD-localized TMEM159/LDAF1 (Chung et al. 2019). By maintaining these contacts, seipin enables continuous delivery of TAGs from the ER to LDs (Salo et al. 2016, 2019). Seipin-null mutations are responsible for the most severe form of congenital generalized lipodystrophy (Magré et al. 2001), characterized by a near-absence of adipose tissue and severe insulin resistance, while dominant mutations cause peripheral neuropathy and other nervous system pathologies. Interestingly, seipin associates at ER-LD contact sites with two key ER enzymes for sphingolipid synthesis, serine palmitoyltransferase and fatty-acid elongase, and negatively regulates their activity (Su et al. 2019). Recent discovery of the involvement of seipin in peroxisome biogenesis further suggests that seipin could have a much broader function beyond LD biogenesis.
Nascent LDs also associate with the ER through a complex between acyl-CoA synthetase FATP1 on the ER and LD-localized diacyglycerol-transferase (DGAT2), which catalyzes the last step of TAG synthesis in eukaryotes, thus coupling lipid synthesis with deposition into LDs at these contact sites (Xu et al. 2012). DGAT2 also recruits mitochondria to LDs (Stone et al. 2009). During nutrient scarcity or cell growth, when there is a high demand for phospholipid biosynthesis for membrane expansion, contact sites between LDs and mitochondria or peroxisomes form to mobilize stored lipids through β-oxidation (Rambold et al. 2015). A missense mutation (Y223H) in DGAT2 has been associated with autosomal dominant Charcot-Marie-Tooth (CMT) disease type 2 (Hong et al. 2016). CMT is the most common inherited peripheral neuropathy associated with mitochondrial dysfunction, characterized by childhood onset of slowly progressive distal muscle weakness with sensory impairment. While type 1 features demyelination, type 2 CMT is characterized by axonal degeneration. It remains unknown as to how DGAT2 is coordinated at distinct contact sites and their contribution to axonal pathology.
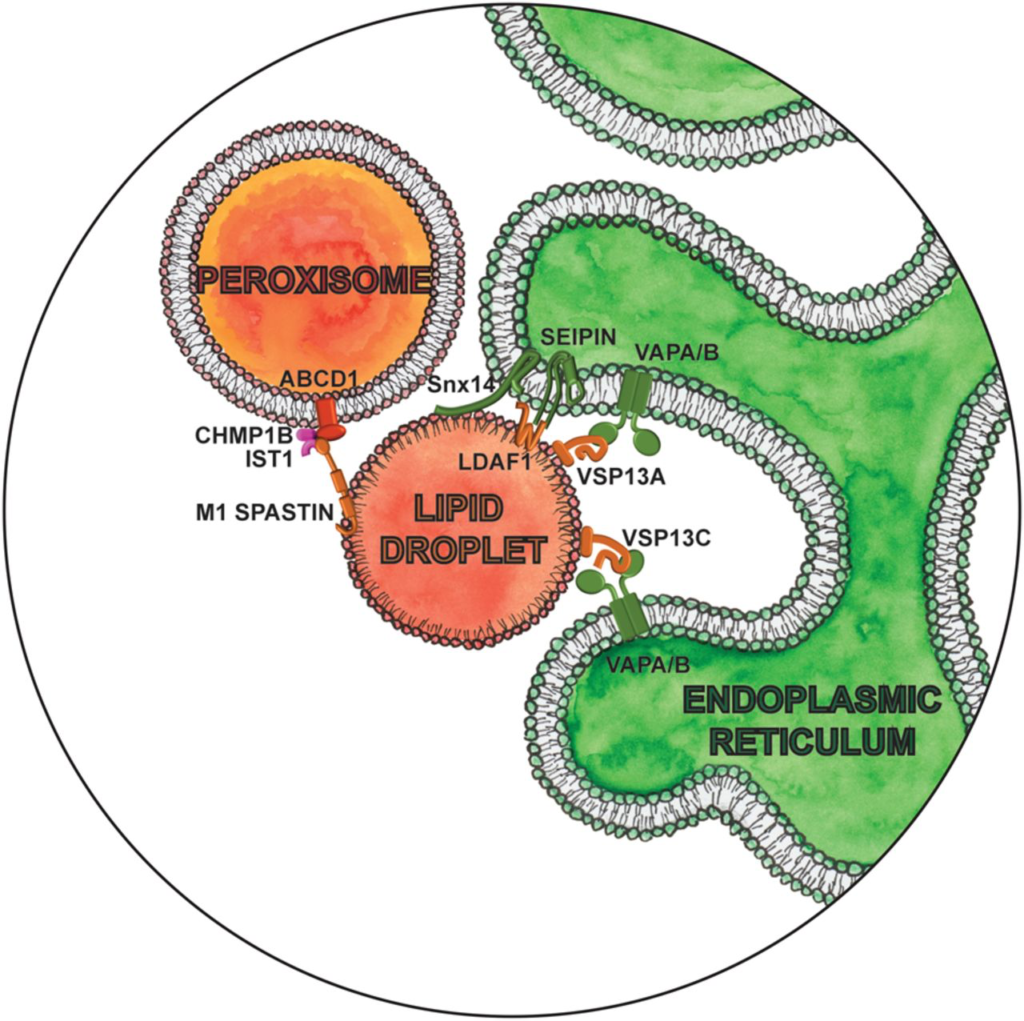
Magnifying glass view of the contact sites between the ER, lipid droplet, and peroxisome. ER-localized proteins are labeled in green. Lipid droplet localized proteins are labeled in orange. Peroxisome proteins are red, and endosome proteins are fushia.
Snx14, like its homologs Mdm1 (in yeast) or Snazarus (in Drosophila), also acts as a tether between the ER and LDs during LD biogenesis (Fig. 3). Snx14 facilitates the conversion of free fatty acids into TAGs and their incorporation into LDs (Datta et al. 2019; Hariri et al. 2019). Disrupting these contacts can result in disease, as exemplified by the loss-of-function mutations in SNX14 that cause spinocerebellar autosomal recessive ataxia 20 (SCAR20) (Bryant et al. 2018). SCAR20 is characterized by severely delayed psychomotor development with poor or absent speech, wide-based or absent gait, coarse facies, and cerebellar atrophy (Thomas et al. 2014). Cells lacking Snx14 display a small decrease in ER-LD contacts and heterogeneity in LD size. Overexpression of seipin cannot rescue LD defects in Snx14-null cells and vice versa (Datta et al. 2019), suggesting they function independently of each other to regulate LD morphology. This is just one of the examples of multiple distinct contacts formed between the same organelles but with nonoverlapping functions. Unesterified cholesterol does accumulate in SCAR20 patient cells (Bryant et al. 2018), indicating impaired lipid metabolism and storage. Disruption of ER-LD contacts as a pathogenic driver in SCAR20 patients is therefore an exciting hypothesis for future studies.
Two members of the VPS13 family of lipid transporters (Li et al. 2020), VPS13A and VPS13C, also localize at contacts between the ER and LDs (Fig. 3; Kumar et al. 2018). Involvement of the two other family members, VPS13B and VPS13D, remains to be determined. However, loss-of-function mutations of any of the four members leads to progressive neurodegeneration: VPS13A is linked with chorea acanthocytosis, a rare disease with clinical features similar to Huntington’s disease (HD) (Rampoldi et al. 2001; Ueno et al. 2001); VPS13C is associated with Parkinson’s disease (PD) (Lesage et al. 2016); mutations of VPS13B cause Cohen syndrome, a rare neurodevelopmental disorder (Kolehmainen et al. 2003); while Vps13D mutations cause spastic ataxia (Gauthier et al. 2018; Seong et al. 2018). Unlike classical lipid transfer proteins, like previously mentioned ORP5 that can transport monomeric lipid molecules between the lipid bilayers, VPS13 family proteins have a channel-like domain that may allow for bulk flow of lipids between organelles at sites of contact (Lees and Reinisch 2020). It is like comparing transport of water in a small plastic bottle to the transport of water through an open dam. Considering that lipids are mainly synthesized in the ER but are primarily stored in LDs, VPS13A and VPS13C seem well suited for the task of mediating bulk lipid transfer at ER-LD contacts. In the absence of VPS13A (Yeshaw et al. 2019), the number of ER-LD contacts are reduced, while the total number of LDs and their mobility are increased. VPS13C depletion does not have an effect on the total number of LDs but reduces their size and increases the free fatty acid content in a cell, probably through increased recruitment of ATGL, a triglyceride lipase, to LD (Ramseyer et al. 2018).
Both VPS13A and VPS13C also act at other distinct contact sites: VPS13A at ER-mitochondria contacts (Fig. 4; Kumar et al. 2018; Yeshaw et al. 2019) and VPS13C at ER-lysosome/late endosome contacts (Fig. 2; Kumar et al. 2018). As with the ER-LD contacts described above, VPS13A and VPS13C interact with the ER-localized VAPA/B proteins at these contact sites. What regulates the recruitment of VPS13A and VPS13C to either LD or to mitochondria and the endolysosomal compartment, respectively, is not fully understood. Yeast has only one Vps13 member (Park et al. 2016), and its localization seems to be regulated by nutrient availability. Organelle-specific adaptors compete for binding to the same conserved six-repeat region, the Vps13 adaptor binding (VAB) domain, enabling Vps13 recruitment to different contact sites (Bean et al. 2018). A similar mechanism could be in place in mammals, as a disease-causing mutation in VPS13D, which maps to this highly conserved VAB domain, which blocks adaptor binding and Vps13 membrane recruitment when modeled in yeast (Dziurdzik et al. 2020). The complexity of interorganelle interactions of this family does not end here. VPS13A and VPS13C also bind endolysosomal GTPase Rab7 (Fig. 4), where depletion of VPS13A, localized to ER-mitochondria and ER-LD contacts, was found to additionally impair lysosomal degradation (Muñoz-Braceras et al. 2019). VPS13C depletion, on the other hand, was also linked with mitochondrial fragmentation, impaired respiration, and increased mitophagy (Lesage et al. 2016). Considering its central role for cell survival, it is not surprising that organelles are highly interconnected in the regulation of sterol and fatty acid homeostasis. Such an interconnected network of organelles could compensate for changes in function between its subcomponents. Indeed, yeast Vps13 can compensate for the absence of ERMES complex-mediated lipid transfer at ER-mitochondria contacts by transferring lipids at contacts between the ER and vacuole (Lang et al. 2015; Park et al. 2016). The Vps13 family and VAPA/B are good examples of proteins that participate in multiple distinct contact sites, thus highlighting the need for more research on how a cell balances physiological processes at different contact sites and how perturbations or compensatory alterations of distinct contact sites may impact the whole network of interorganelle communication, specifically leading to one disease but not another.
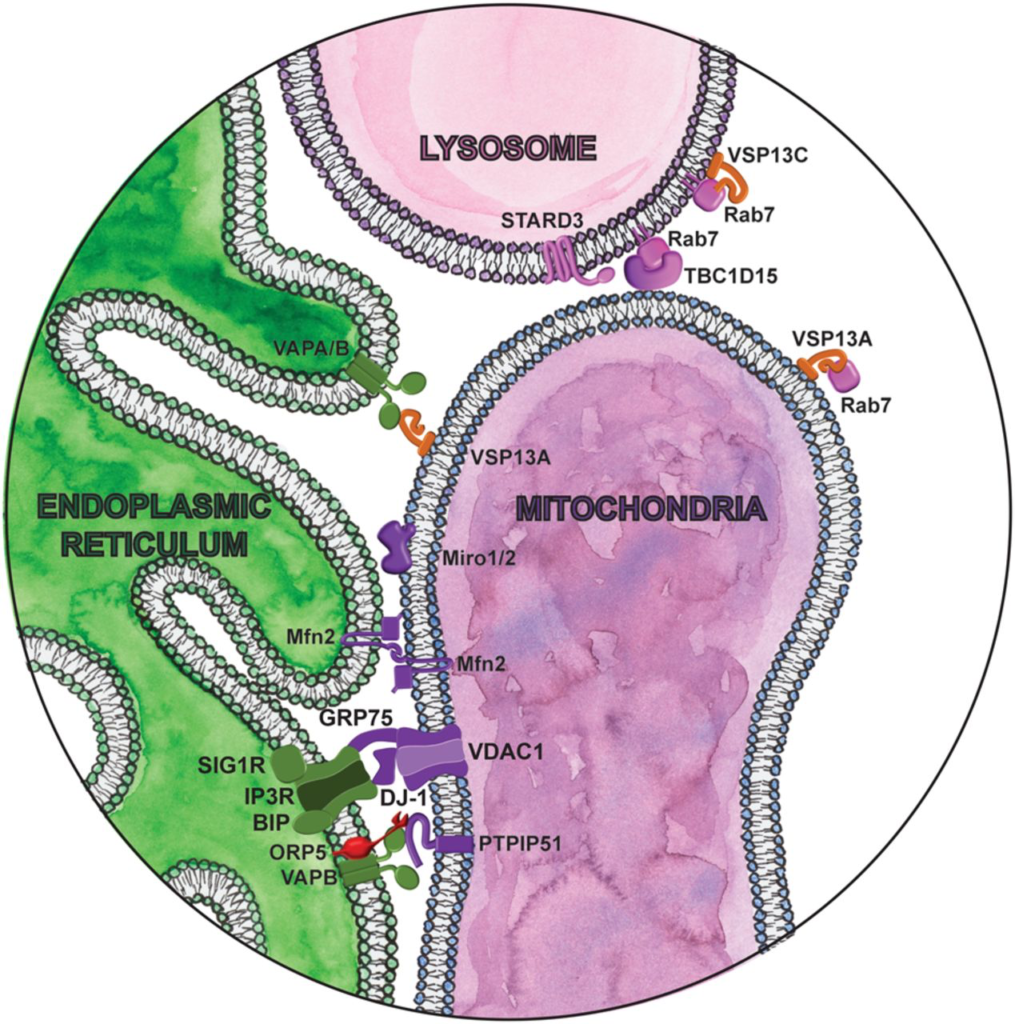
Magnifying glass view of the contact sites between the ER and mitochondria. ER-localized proteins are labeled in green. Lipid droplet localized proteins are labeled in orange. Lysosome proteins are labeled in pink. Cytosolic lipid transfer proteins are labeled in red. Mitochondria proteins are labeled in purple.
LDs form contact sites not only to store or mobilize lipids but also to eliminate toxic lipid species. Trafficking of fatty acids through LD-peroxisome contacts prevents accumulation of peroxidated lipids following oxidative stress. Under basal conditions as well as during oxidative stress (Chang et al. 2019), the M1 isoform of spastin regulates these contacts and the transfer of fatty acids from LD to peroxisomes (Fig. 3). Spastin is a microtubule severing protein whose mutations are the most common cause of hereditary spastic paraplegias (HSP), characterized by progressive weakness and spasticity of the lower limbs (Fonknechten et al. 2000). A spastin mutation associated with HSP reduces LD-peroxisome contacts and results in the accumulation of peroxidated lipids (Chang et al. 2019). While LD-peroxisome contacts have yet to be measured in HSP patient cells, enhanced levels of lipid peroxidation have been observed (Chang et al. 2019). Spastin mediates LD-peroxisome contacts through interaction with peroxisomal fatty acid transporter ABCD1 and components of the endosomal sorting complexes required for transport (ESCRT) III machinery, Ist1 and CHMP1B (Chang et al. 2019). It is presumed that ESCRT III machinery is recruited for their membrane deformation abilities to extract lipids from the LD core. Interestingly, the M1 isoform of spastin also localizes at ER-endosome contacts required for endosomal fission through its interaction with the same ESCRT III protein, Ist1 (Allison et al. 2017). Endosomal fission is essential to separate regions destined for lysosomal degradation from carriers to be recycled. Defective endosomal sorting and abnormal lysosomal function were found in primary cortical neurons from a spastin-HSP mouse model, patient fibroblasts, and induced pluripotent stem cell (iPSC)-derived patient neurons. How the function of spastin and Ist1 at two distinct sites is coordinated, as well as how they contribute to HSP pathology, is as yet unclear, requiring better understanding of the network of interorganelle communication as a whole. Spastin may play a role in reorganizing the ER along microtubules, thereby influencing the sites of endosome fission and LD biogenesis (Allison et al. 2019; Arribat et al. 2020). It is interesting to note that ABCD1 mutations are causative for adrenoleukodystrophy, a neurodegenerative disease that symptomatically often mimics HSP (Shaw-Smith et al. 2004), suggesting that an underlying defect in HSP may be peroxisomal dysfunction due an imbalance in spastin-mediated contact sites.
Mitochondrial contact sites
Mitochondria are a key organelle in eukaryotes, sitting at the interface of cellular metabolism, energy production, and cell death. Membrane contact sites, in particular those between the ER and mitochondria called MERCs, have been implicated in various aspects of mitochondrial function, like mitochondrial morphology, dynamics, calcium homeostasis, and mitophagy. While the term MAM previously used in this review refers to the mitochondrial-associated membrane fraction of the ER, the term MERCs refers to the sites of contact between the ER and mitochondria. There is growing evidence that perturbation of MERCs is a major driver of aging and various age-related pathologies (Moltedo et al. 2019). While we focus much of this section on MERCs, we also highlight contact sites between mitochondria and other organelles with pathological relevance.
Mitofusin 1 and 2 (Mfn1 and Mfn2) are tethering proteins critical for mitochondrial fusion in response to metabolic and environmental cues (Chen et al. 2003). Mitochondria fusion affects their morphology, dynamics, distribution, quality control, and function, which in turn critically influences cellular fitness. Mfn2 was also one of the first described contact site proteins (de Brito and Scorrano 2008), localized at MERCs, where it interacts both homotypically and heterotypically with mitochondrial-localized Mfn1 and Mfn2 (Fig. 4; de Brito and Scorrano 2008). How Mfn2 influences MERCs is somewhat controversial, as Mfn2 was reported to both promote (de Brito and Scorrano 2008; Naon et al. 2016; Basso et al. 2018; McLelland et al. 2018) and inhibit (Cosson et al. 2012; Filadi et al. 2015) MERCs, highlighting the physiological complexity of these contacts. Like DGAT2, mutations in MFN2 are associated with CMT disease type 2 (Züchner et al. 2004). In fibroblasts from CMT patients with several different MFN2 mutations, the distance between the ER and mitochondria is greater, while the length of contacts between these organelles is increased (Larrea et al. 2019). Cells with the Mfn2 R364W mutation, which results in a severe form of CMT disease, show an increase in synthesis of phosphatidylethanolamine (PE) and phosphatidyserine (PS), as well as cholesterol esters, both processes that occur at MERCs. However, these functional changes are not observed in patients with a less severe form of CMT. As Mfn2 mutations in CMT are primarily heterozygous dominant, wild-type protein exists in these cells, which may dampen any deleterious effects of the mutant protein. It is also unclear how exactly these mutations affect the ability of Mfn2 to interact with wild-type Mfn1 or Mfn2. Interestingly, a role in ER-mitochondria contacts in inhibiting mitophagy was recently shown (Basso et al. 2018; McLelland et al. 2018), suggesting that turnover of damaged mitochondria may be affected in CMT.
Mitochondrial contacts with other organelles also appear to be linked with CMT disease. Mitochondria dynamically form contacts between themselves and with lysosomes. Mitochondrial-lysosome contacts are regulated by the endolysosomal GTPase Rab7 (Fig. 4; Wong et al. 2018). GTP hydrolysis by Rab7 promotes mitochondrial-lysosome untethering, as expression of the GTP-bound Rab7 mutant (Rab7 Q67L) or TBC1D15 mutants lacking Rab7 GAP activity (D397A and R400K) increases the frequency and duration of these contacts (Wong et al. 2018). Mitochondrial-lysosome contacts are distinct from degradative pathways, such as mitophagy or the formation of mitochondria-derived vesicles, and do not involve the bulk transfer of proteins between the organelles. Whether these contacts permit the transfer of other material such as ions or lipids is unknown. Mitochondrial-lysosome contacts, however, do appear to regulate intermitochondrial contacts (Wong et al. 2019). Like other contact sites, intermitochondrial contacts are dynamic and undergo frequent tethering and untethering. Mfn1 and Mfn2 are required for the formation of intermitochondrial contacts, while recruitment of Drp1 oligomers and the formation of a mitochondrial-lysosome contact site marks intermitochondrial contact sites for untethering. GTP hydrolysis by either Drp1 or Rab7 promotes intermitochondrial untethering (Wong et al. 2019). Expression of a CMT type 2-associated Rab7 mutation (V126M) or Mfn2 mutation (T105M) results in prolonged intermitochondrial contact (Wong et al. 2019). How strengthening these contacts affects the Mfn2-mediated MERCs discussed earlier has not been explored. Surprisingly, the independent CMT type 2-associated TRPV4 R269H mutation with no direct role in contact site formation also prolongs intermitochondrial contacts (Wong et al. 2019), suggesting that disruption of intermitochondrial contacts may contribute to pathogenesis. Considering that TRPV4 is a nonselective calcium permeant cation channel and the R269H mutation increases constitutive function (Klein et al. 2011), Ca2+ transport at these sites might be also dysregulated and contribute to the disease. Interestingly, all CMT-associated mutations we have discussed in relation to contact sites produce the type 2 axonal, not type 1 demyelinating, phenotype of this disease, suggesting that neurons are selectively vulnerable to these disruptions. This raises the intriguing possibility that different cell types may rely on different subsets of contact sites or require different levels of interorganelle communication.
Mitochondrial contact sites are essential for Ca2+ homeostasis and buffering cytosolic Ca2+ levels. Both mitochondria and the ER are major Ca2+ stores in the cell. Within mitochondria, Ca2+ levels regulate ATP production (Tarasov et al. 2012), and mitochondrial Ca2+ overload can trigger apoptosis (Pinton et al. 2008). A complex between the mitochondrial localized voltage-dependent anion channel 1 (VDAC1), the ER localized IP3 receptor (IP3R), and the ER chaperone Grp75 facilitates Ca2+ transfer from the ER to mitochondria (Szabadkai et al. 2006). Knockdown of Grp75 abolishes the coupling between these ER and mitochondrial ion channels and prevents Ca2+ transfer to mitochondria.
The σ1 receptor (Sig1R), an ER chaperone with cytoprotective activity (Mori et al. 2013), localizes to MERCs and can stabilize IP3R. Under basal conditions, Sig1R interacts with calcium binding protein BiP, but in response to ER or mitochondrial stress as well as IP3R activation (Hayashi and Su 2007), Sig1R dissociates from BiP and interacts with IP3R, preventing its degradation (Hayashi and Su 2007). Depletion of Sig1R attenuates Ca2+transfer to mitochondria, while leaving Ca2+ release to the cytosol intact, suggesting that Sig1R may affect the Grp75-VDAC-IP3R complex and MERCs specifically, beyond simply stabilizing IP3R. Mutations in the SIGMAR1 gene encoding Sig1R have been linked to juvenile-onset (Al-Saif et al. 2011) and adult-onset (Ullah et al. 2015) forms of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD) (Luty et al. 2010), and distal hereditary motor neuropathy (Gregianin et al. 2016). These studies report that Sig1R mutations result in ER stress, disturbed intracellular Ca2+ signaling, and defects in mitochondrial dynamics and transport. While these initial studies did not examine MERCs or Grp75-VDAC-IP3R, a subsequent report suggests that MERC disruption in motor neurons is common to ALS-linked mutations in either Sig1R or superoxide dismutase (SOD1) (Watanabe et al. 2016), the latter being the most common cause of ALS, which itself does not function at these contacts. Further arguing for a role of MERC disruption as a pathogenic driver of neurodegeneration, stimulation of Sig1R with the agonist Pre-084 prevents axonal degeneration in motoneurons in an SOD1 mutant mouse model of ALS (Mancuso et al. 2012). What is not clear from these studies is whether rescuing Ca2+transfer through IP3R is sufficient to prevent neurodegeneration or whether the protective effect of Sig1R agonists is mediated by additional targets of Sig1R’s chaperone function or MERC-associated processes more broadly. Beyond motor neuron disease, Sig1R polymorphisms have been identified as a risk factor for Alzheimer’s disease (AD) (Fehér et al. 2012), and Sig1R agonists are protective in a cellular model of Huntington’s disease (HD) (Hyrskyluoto et al. 2013), making Sig1R an attractive therapeutic target for multiple neurodegenerative diseases. We discuss additional implications of Sig1R and MERCs in HD and AD below.
More recently, DJ-1 has also been reported to interact with the Grp75-VDAC-IP3R tethering complex to stabilize MERCs and facilitate Ca2+ transfer. In the absence of DJ-1, Grp75, VDAC1, and IP3R interact less, resulting in smaller MERCs, decreased mitochondrial Ca2+ uptake, and decreased ATP production (Liu et al. 2020). Overexpression of DJ-1 can also enhance mitochondrial Ca2+ uptake (Ottolini et al. 2013). Mutations in DJ-1 have been linked to a rare form of early-onset familial Parkinson’s disease (PD) (Bonifati 2003; Repici and Giorgini 2019). While DJ-1 also functions in oxidative stress responses (Canet-Avilés et al. 2004) and mitophagy (Krebiehl et al. 2010), at least some DJ-1 PD mutations appear to selectively affect mitochondrial Ca2+ dynamics. Specifically, the D149A, but not the M26I, DJ-1 mutation increases mitochondrial Ca2+ load, leading to mitochondrial fragmentation and ROS production (Strobbe et al. 2018). Conversely, the M26I, but not the D149A, mutation impairs mitophagy. While this study did not measure MERC abundance or stability of the Grp75-VDAC-IP3R complex, the increase in mitochondrial Ca2+ suggests that the D149A mutation may enhance these interactions.
The multiple functions of mitochondria, including ATP production, Ca2+ flux, reactive oxygen species (ROS) formation, and apoptosis regulation can be affected by mitochondrial dynamics. Miro1 and Miro2 are Rho-GTPases best known for their role in mitochondrial transport (López-Doménech et al. 2018). The only yeast homolog, Gem1, is an integral part of the ERMES complex at sites of contact between the ER and mitochondria, responsible for mitochondrial inheritance. Likewise, mammalian Miro localizes to MERCs (Fig. 4; Kornmann et al. 2011), where it facilitates mitochondrial Ca2+uptake from the ER (Lee et al. 2018). Knockdown of Miro in dPINK1 mutant flies, a Parkinson’s disease model, rescues excess Ca2+ transfer at contacts and prevents neurodegeneration (Wang et al. 2011). In mammalian cells, Miro1/2 double knockout reduces contact site number and impairs IP3R-dependent Ca2+ transfer (Modi et al. 2019). Miro also anchors the mitochondrial MICOS complex to the kinesin adaptor TRAK1/milton (Modi et al. 2019). In the absence of this cytoskeletal tether in Miro1/2 double KO cells, the MICOS complex is unevenly distributed across the surface of the mitochondria, disrupting the cristae structure (Modi et al. 2019) Genetic interaction studies in yeast identified strong interactions between ERMES, MICOS, and Gem1 (Hoppins et al. 2011). As the MICOS complex is required for respiratory chain function and oxidative phosphorylation (Friedman et al. 2015), mutation of Miro or its aberrant degradation by PINK1/parkin may explain the disturbances in bioenergetics observed in PD. In PD patient-derived fibroblasts and iPSC-derived neurons, Miro mutations change its retention at MERCs and MERC abundance—mutations that decrease its retention (T351A and T610A) decrease MERC number, while those that increase Miro retention at MERCs (R272Q) increase MERC number (Berenguer-Escuder et al. 2019, 2020). Regardless of the effect on MERC abundance, these perturbations cause similar functional defects: leakage of Ca2+ into the cytoplasm, mitochondrial fragmentation, and impaired mitophagy. It was suggested that this apparent discrepancy may reflect the different cell types used in these studies. In our view, these results also suggest that an increase in the physical contact between organelles does not necessarily mean the contacts are functional, or physiologically optimal, and highlight the need to determine how disease-associated mutations affect both the structural and functional aspects of contact sites.
VAPB is an ER-localized protein involved in contact sites between the ER and multiple different organelles (Figs. 2–5). In MERCs, VAPB interacts with the protein tyrosine phosphatase interacting protein 51 (PTPIP51), an outer mitochondrial membrane protein with apoptosis-inducing function (Fig. 4; De Vos et al. 2012). Modulating the levels of either protein by overexpression or siRNA knockdown increases or decreases MERCs, respectively (Stoica et al. 2014). VAPB-PTPIP51 contacts were also found at synapses, where synaptic activity increases MERCs. Knockdown of either VAPB or PTPIP51 inhibits synaptic activity, including alterations to synaptic vesicle release and dendritic spine numbers. VAPB-PTPIP51 interaction is dysregulated in FTD (De Vos et al. 2012; Stoica et al. 2014; Gómez-Suaga et al. 2018). Mutations in VAPB also cause dominantly inherited familial forms of motor neuron disease including ALS and spinal muscular atrophy (SMA) (Nishimura et al. 2004). The ALS associated VAPB mutation P56S increases interaction between VAPB with PTPIP51, recruiting more VAPB to MERCs, and inducing mitochondrial clustering (De Vos et al. 2012). This in turn enhances mitochondrial Ca2+uptake. Disturbances in Ca2+ homeostasis and in the Ca2+ buffering capacity of mitochondria is a common feature in ALS and may contribute to the excitotoxicity of motor neurons (Grosskreutz et al. 2010).
Two lipid transfer proteins, ORP5 and ORP8, interact independently with both VAPB and PTPIP51 at MERCs upon phosphatidylserine (PS) binding (Galmes et al. 2016), linking lipid transfer to VAPB-PTPIP51 contact sites. Overexpression of ORP5 increases these contacts, as shown for VAPB and PTPIP51, while depletion of ORP5/ORP8 leads to defects in mitochondrial morphology and respiration. ORP5 and ORP8 were recently found to transport PS from its site of synthesis in the ER to mitochondria, where PS is converted to phosphatidylethanolamine (PE), an essential phospholipid for maintenance of mitochondrial morphology and hence respiratory function (Rochin et al. 2019). ORP5 also plays an additional role in regulating mitochondrial Ca2+ uptake at ER-mitochondria contact sites (Rochin et al. 2019). Whether and how these observed functions are interconnected remains to be determined.
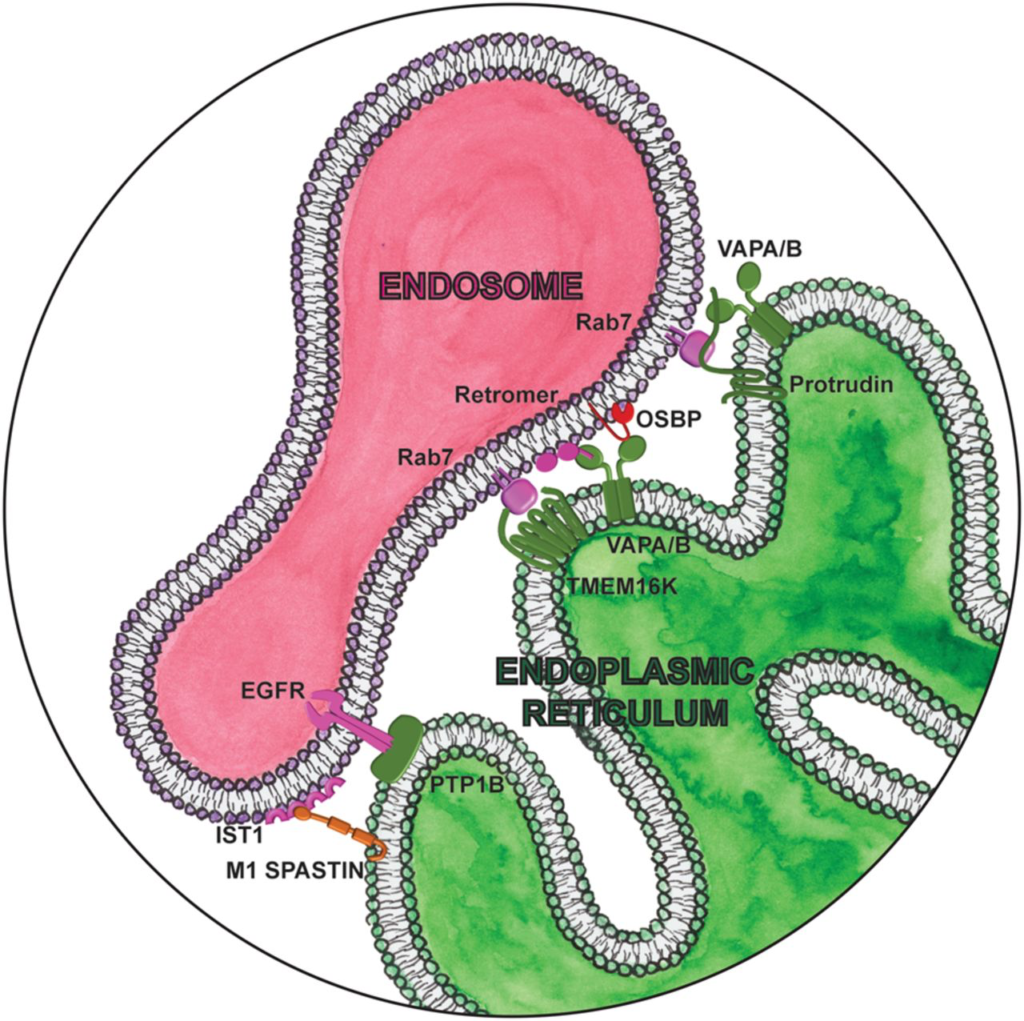
Magnifying glass view of the contact sites between the ER and endosome. ER-localized proteins are labeled in green. Lipid droplet localized proteins are labeled in orange. Cytosolic lipid transfer proteins are labeled in red.
VAPB-mediated contacts also play a role in autophagy regulation. Increased VAPB-PTPIP51 interactions impair, whereas siRNA depletion of either protein stimulates, autophagosome formation. An artificial tether returns autophagy levels to baseline in the absence of VAPB or PTPIP51, while preventing mitochondrial Ca2+ uptake abrogates this effect. This suggests that this functional role of VAPB-PTPIP51 contacts is essential to autophagy regulation (Gomez-Suaga et al. 2017). Another study found that VAPA/B are recruited to sites of autophagosome formation on the ER, where they directly interact with multiple autophagy (ATG) proteins. VAPB depletion or the P56S mutation impairs this interaction and inhibits autophagy (Zhao et al. 2018). Whether these two roles of VAPB in autophagy regulation are mutually exclusive is unclear. The repertoire of VAPA/B interactions and the variety of functions they regulate again exemplifies the interconnectivity of cellular processes, highlighting the need for better understanding of the network of interorganellar communication as a whole. In contrast, the redundancy of proteins capable of mediating distinct contact sites like MERCs also highlights the need to disentangle their functional relationships. One exciting open question is whether the function of one organelle or subnetwork takes precedence over the functioning of another in conditions of limited resources or under stress.
Contact sites in the endolysosomal system
Endolysosomal compartments integrate external signaling with the internal metabolic status of the cell. Interorganelle communication regulates various aspects of endolyososomal function, including receptor signaling, protein and lipid sorting, endosomal maturation, and acidification that is essential for proper degradation in the lysosome. Dysfunction in this pathway is a feature of many neurodegenerative diseases (Malik et al. 2019) and has been linked with the disruption of specific contact sites in recent studies.
Receptor tyrosine kinases (RTKs) are cell surface receptors for growth factors, cytokines, and hormones, playing an important role in a variety of cellular processes including growth, motility, differentiation, and metabolism. Signaling from RTKs is temporally and spatially regulated by progression through the endocytic pathway. The ER-localized protein tyrosine phosphatase 1B (PTP1B) dephosphorylates and inactivates several RTKs, including the insulin receptor, leptin receptor, and EGFR. PTP1B interacts with EGFR at membrane contacts between EGFR in endosomes and the ER (Fig. 5; Eden et al. 2010), promoting sequestration of EGFR into intraluminal vesicles (ILVs). Whether PTP1B similarly regulates insulin or leptin receptors at contact sites is an exciting possibility that remains to be evaluated. Another PTP1B substrate is PTPIP51 (mentioned above), where the extent of their interaction is strongly reduced in an animal model of obesity (Bobrich et al. 2011). Whether MERC abundance influences insulin sensitivity or how insulin resistance affects these contacts is unclear (Keenan et al. 2020). However, disruption of ER-mitochondria communication seems to be an early event in the development of insulin resistance, preceding mitochondrial dysfunction and defective insulin signaling (Tubbs et al. 2018). Insulin signaling regulates longevity, and the development of insulin resistance is associated with a number of age-related pathologies including metabolic syndrome, type 2 diabetes, and Alzheimer’s disease (AD) (Roberts et al. 2013; Vieira et al. 2018). In fact, PTP1B inhibitors are emerging as potential therapeutics for neurodegenerative disease (Ricke et al. 2020). Understanding the effect of these inhibitors on MERCs will be critical.
Proper progression through the endocytic pathway requires endosomal content to be sorted depending whether it is destined for recycling to the plasma membrane, retrograde transport to the trans-Golgi, or degradation in the lysosome. We recently found endosomal sorting is regulated by the endoplasmic reticulum localized lipid scramblase TMEM16K (Bushell et al. 2019) at ER-endosome contact sites (Fig. 5; Petkovic et al. 2020). TMEM16K forms contact sites with endosomes by binding to active GTP-bound Rab7. Similarly to the only TMEM16 protein in yeast, Ist2p, that mediates ER-plasma membrane contact sites by directly binding plasma membrane-specific phosphatidylinositol-(4,5)-bisphosphate [PtdIns(4,5)P2] via its C terminus (Fischer et al. 2009), TMEM16K also binds endolysosomal phosphatidylinositols through its N terminus. These TMEM16K-mediated contacts between ER and endosomes are physiologically relevant, as cells lacking TMEM16K exhibit dysfunctional endosomal sorting, leading to defects in endosomal retrograde transport and downstream endolysosomal acidification. Mutations in ANO10, encoding TMEM16K, are causative for autosomal recessive spinocerebellar ataxia (SCAR10), manifesting with gait and appendicular ataxia, dysarthria, ocular movement anomalies, increased deep tendon reflexes, and progressive cognitive decline. (Vermeer et al. 2010). TMEM16K cellular defects could be rescued by wild-type TMEM16K. However, three different human disease point mutants of TMEM16K causing spinocerebellar ataxia (F171S, F337V, and D615N) were not able to rescue cellular defects, and only one (F171S) was able to still interact with Rab7, suggesting that impairment of the endosomal pathway could be a contributing factor in the development of the SCAR10 pathology. How TMEM16K supports endosomal sorting is still unclear, but we hypothesize that endosomal sorting could be enabled by modulating the lipid environment in trans at sites of contact between organelles (Petkovic et al. 2020). Intriguingly, Ist2 in yeast interacts with soluble lipid transfer proteins to mediate lipid transport at contact sites (D’Ambrosio et al. 2020). VAPA/B also support endosomal sorting and transport at ER-endosome contacts by interacting with retromer components and lipid transport protein OSBP. These contacts regulate PtdInsI4P levels on endosomes and subsequent WASH-dependent actin nucleation (Dong et al. 2016). While there could be coordination of TMEM16K and VAPA/B contact sites in mediating endosomal retrograde trafficking, they seem to regulate endosomal sorting independently from each other (Petkovic et al. 2020).
Once sorted, endosomes are transported to their destination. ER-endosomal contact sites positively regulate plus-end microtubule endosomal transport via protrudin, a process implicated in neurite outgrowth. Highlighting its physiological importance, mutations in protrudin are associated with a dominant form of hereditary spastic paraplegia (SPG33) (Mannan et al. 2006). Protrudin is an ER protein that interacts with VAPA/B and binds endosomal PI3P and active GTP-bound Rab7 at ER-endosome contacts (Fig. 5). Interaction with VAPs is required for ER retention of protrudin, as siRNA knockdown of VAPA results in protrudin mislocalization, as well as inhibition of NGF-induced neurite outgrowth in PC12 cells. The protrudin-VAP-Rab7-PI3P complex associates with kinesin-1 and promotes its association with FYCO1, a Rab7 effector, to mediate microtubule transport of late endosomes (Raiborg et al. 2015). Overexpression of protrudin causes the dispersion of late endosomes toward the cell periphery, while depletion perturbs transport, inhibits neurite outgrowth, and stimulates autophagy (Hong et al. 2017). Recently, it was found that increasing protrudin expression enables regeneration of adult axons both in vitro in primary cortical neurons and in vivo in the injured adult optic nerve (Petrova et al. 2020). Considering the energy expenditure associated with neurite growth, it is understandable that protrudin-mediated anterograde transport is regulated by the metabolic status of the cell. Amino acids stimulate FYCO1 recruitment to late endosomes and promote contacts between FYCO1 and protrudin (Hong et al. 2017). Likewise, CPT1C, a sensor of malonyl-CoA with a key role in fatty acid synthesis, promotes anterograde transport by enhancing protrudin-mediated recruitment of kinesin-1. Mutations in CPT1C have also been associated with hereditary spastic paraplegia (Rinaldi et al. 2015). Furthermore, recent studies suggest that mitochondria are also recruited at these ER-late endosome contacts, forming a three-way contact through interaction with PDZD8 (Guillén-Samander et al. 2019; Elbaz-Alon et al. 2020; Shirane et al. 2020). While the ERMES complex seems to exist only in yeast, PDZD8 was suggested as a putative mammalian ortholog of the ERMES complex at ER-mitochondria MCSs where it was reported to participate in calcium regulation (Hirabayashi et al. 2017). However, PDZD8 also interacts independently with both protrudin and active GTP-bound Rab7 and transfers lipids at these sites of contact to enable endosomal maturation (Shirane et al. 2020). To understand how this may drive HSP pathogenesis, it will be important to determine how all these pieces fit together to connect the metabolic status of the cell with transport.
Contribution of dysfunctional interorganelle communication to aging and neurodegeneration
Even if the original insult, to our current knowledge, does not directly affect contact sites per se, contact site perturbations could be a contributing factor in aging and disease progression by allowing age- or disease-related dysfunction in one organelle or process to spread throughout the interorganelle communication network (Fig. 6). One of the first observations that contact sites could be affected in neurodegenerative disease was made some 20 years ago in the mnd mouse model of neuronal ceroid lipofuscinosis (Vance et al. 1997), an inheritable lysosomal storage disorder due to a spontaneous mutation in the CLN8 gene (Ranta et al. 1999). In this study, levels and activity of phosphatidylethanolamine N-methyltransferase (PEMT2) and two other key enzymes for phospholipid synthesis, normally found at MERCs, were greatly reduced, suggesting the functionality of this contact is perturbed (Vance et al. 1997). CLN8 was recently shown to be an ER export receptor for lysosomal proteins, whose deficiency leads to depletion of soluble enzymes in the lysosome and impaired lysosome biogenesis (di Ronza et al. 2018). Altered CLN8 function was linked with progressive ER stress (Galizzi et al. 2011), likely due to accumulation of protein in the ER. ER stress may explain how CLN8 mutations disrupt contact sites, as ER stress increases MERCs (Bravo et al. 2011) and provokes Ca2+ leak from the ER and increased mitochondrial Ca2+ uptake (Chami et al. 2008). Indeed, age-dependent progressive deficiency of mitochondrial Ca2+ clearance (Kolikova et al. 2011) was observed in this mouse model, even before the onset of behavioral symptoms. This further suggests that MERC perturbation takes place early in the development of pathology. How exactly mutations in CLN8 lead to ER-mitochondria contact site dysfunction and to what degree this contributes to the progression of pathology remains to be determined. Interestingly, CLN8 has been also reported to interact with VAPA (Passantino et al. 2013), further suggesting disruption of contact sites in the progression of this disease.
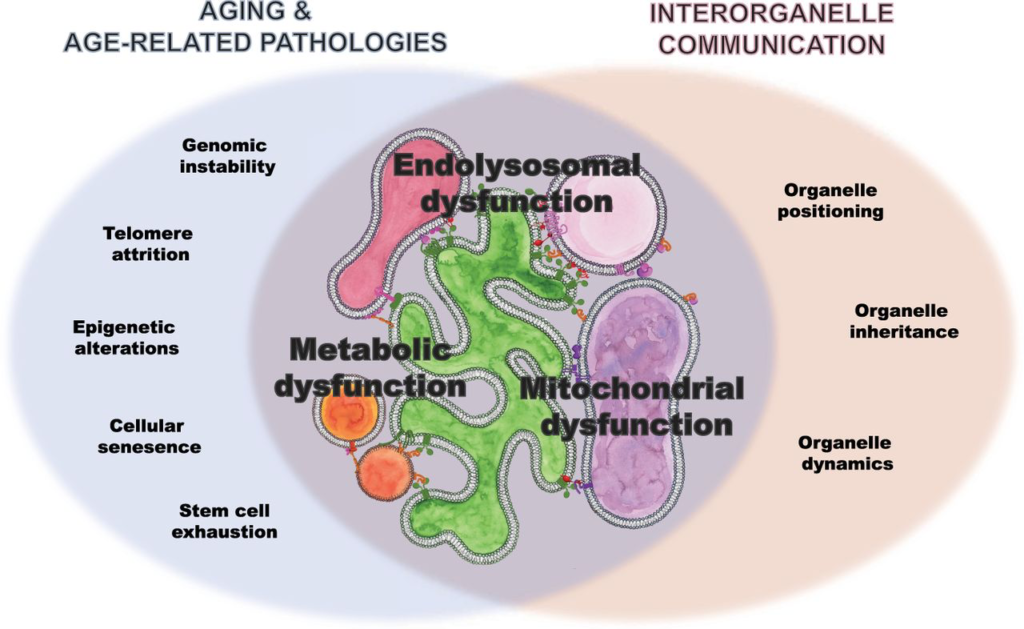
Venn diagram showing the overlap of processes implicated in age and age-related pathologies as well as supported by interorganelle communication, on which we focused in this review: dysfunction in lipid metabolism, mitochondrial function, and endolysosomal system.
While the VAPB and Sig1R mutations discussed in “Contact Sites Directly Linked with Aging and Neurodegeneration” represent only a small number of ALS/FTD cases, the more commonly mutated genes, TARDP43 (TDP43) and FUS, may also interfere with MERCs. Overexpression of TDP43 or FUS, both in culture and in transgenic mice, decreases binding between VAPB and PTPIP51, as well as MERC abundance, resulting in leakage of Ca2+ into the cytosol and decreased ATP production (Stoica et al. 2014, 2016). This appears to be an indirect effect, as neither TDP43 nor FUS bind either VAPB or PTPIP51, but rather they activate glycogen synthase kinase 3β (GSK3β). Inhibition of GSK3β promotes the VAPB-PTPIP51 interaction and increases MERC abundance (Stoica et al. 2016). GSK3β presumably regulates the VAPB-PTPIP51 interaction through phosphorylation, although this has not been directly demonstrated. Disease-associated TDP43 or FUS mutations disrupt the VAPB-PTPIP51 interaction to a similar degree, as does overexpression of the wild-type protein, so it is not clear what significance this regulation may have in disease pathogenesis. Whether these mutations also affect the Sig1R-IP3R interaction and Ca2+ transfer at those sites is unclear. Evidence of disrupted MERCs as a common feature in ALS/FTD patients is also still lacking.
In primary fibroblasts obtained from familial and sporadic AD patients, MERCs are expanded (Area-Gomez et al. 2012), suggesting perturbation of MERCs may contribute to AD pathology. Likewise, increased cytosolic Ca2+ and mitochondrial depolarization was reported (Gautier et al. 2016), indicating that increased MERCs perturb mitochondrial function. Several studies have further reported that MERCs regulate amyloid β-peptide metabolism and, conversely, that amyloid β-peptide can influence the number of these sites (Schreiner et al. 2015). Presenilin 2 (PS2), one component of the γ-secretase complex that participates in amyloid β processing, can also regulate MERCs. Expression of wild type or the AD-associated PS2 mutants that abolish γ-secretase activity increases MERCs and mitochondrial Ca2+ uptake (Zampese et al. 2011). However, it is as yet unclear whether PS2 affects MERCs directly, through amyloid β production or processing of some other substrate. Analysis of brain biopsies from patients with idiopathic normal pressure hydrocephalus, a neurological disorder often comorbid with AD, found that almost fifty percent of mitochondria were in contact with at least one stretch of ER. The number of these contact sites positively correlates with dementia, cognitive impairment, and age of the patients (Leal et al. 2018), thereby implicating contact site dysfunction with pathology progression. ER-mitochondrial communication and MAM function, measured by the synthesis of phospholipids and cholesterol esters, respectively, are increased significantly in cells treated with conditioned-media containing apolipoprotein E4 (ApoE4), a risk factor for AD, but not ApoE3 (Tambini et al. 2016). Similar observations have led Schon and Area-Gomez (2013) to propose that dysregulation of MERCs in AD may explain the pathological disturbances that the amyloid hypothesis cannot, including dysregulation of Ca2+, cholesterol and phospholipid metabolism, and mitochondrial function.
Mitochondrial fragmentation and disruption of Ca2+ buffering and ATP production are also features of Huntington’s disease (HD) (Carmo et al. 2018). Recent studies suggest that MERCs themselves may be disrupted in HD. MERCs are decreased in primary striatal cultures from the R6/1 HD mouse model (Cherubini et al. 2020). Similarly, expression of multiple MERC tethers including Grp75, IP3R, and Mfn2 is decreased in striatal tissue from R6/1 and HdhQ7/Q111 mice, as well in HD patient samples. Preventing Drp1-mediated mitochondrial fission using the small molecule inhibitor Mdivi-1 rescues MERC abundance, superoxide production, and Ca2+ transfer between the ER and mitochondria. Indeed, Drp1 hyperactivation is observed in many HD mouse models resulting in mitochondrial fragmentation. In HD patient-derived, but not healthy subject-derived, iPSCs, the ATPase family AAA-domain containing protein 3A (ATAD3A) aberrantly interacts with Drp1 at points of contact between the mitochondrial inner and outer membranes (Zhao et al. 2019). ATAD3A oligomers, which promote Drp1 activity, are present at higher levels in the brains of both R6/2 mice and HD patients. A peptide that interferes with the ATAD3A-Drp1 interaction rescues mitochondrial defects and cell death in HD iPSCs, as well as the neuropathology and associated behavioral defects in HD mice. Given the relationship between MERCs and Drp1-mediated fission, examination of these contact sites in this context is merited. These studies and the Sig1R agonist study (Hyrskyluoto et al. 2013) referenced in “Contact Sites Directly Linked with Aging and Neurodegeneration” suggest that restoring at least one MERC tethering complex is protective in HD models. What is as yet unclear is how mutant huntingtin disrupts them in the first place and whether these treatments affect the other tethering complexes.
In addition to the studies on DJ-1 at MERCs that we discuss above, there is a growing literature linking alterations in contact sites with Parkinson’s disease (PD). In particular, the E3 ubiquitin ligase parkin, and its regulatory kinase PINK1 (PTEN-inducible kinase 1), both linked to familial PD, appear to regulate several interorganelle contacts, including MERCs. Parkin and PINK1 target a number of mitochondrial proteins such as Mfn1/2 for degradation (Ziviani et al. 2010; Glauser et al. 2011; Sarraf et al. 2013; Gautier et al. 2016; Basso et al. 2018). Depletion of parkin decreases overall Mfn2 ubiquitination (Basso et al. 2018) and specifically increases the MAM-localized pool of Mfn2 (Gautier et al. 2016). Loss of function of PINK1 or parkin results in mitochondrial fragmentation and disrupted cristae structure, reduced mitochondrial membrane potential, and decreased ATP production, contributing to both neurodegeneration and an overall reduction in life span (Clark et al. 2006; Park et al. 2006; Exner et al. 2007; Yu et al. 2011). While the role of PINK1/parkin in the degradation of damaged mitochondria through mitophagy is well documented (Eiyama and Okamoto 2015), the ability of parkin to ubiquitinate Mfn2 has generated interest in how parkin loss of function affects MERC abundance. Several studies report that parkin knockdown decreases MERCs as well as mitochondrial Ca2+transients (Clark et al. 2006; Park et al. 2006; Exner et al. 2007), while others found that in PARK2-patient-derived fibroblasts and parkin knockout mouse embryonic fibroblasts, there is an increase in ER-mitochondrial proximity and enhanced IP3R-mediated Ca2+transfer to mitochondria. Further, these effects can be mitigated by Mfn2 knockdown. Additional studies observe an increase in MERCS in mouse models of PD with knockout of PINK1 or parkin, as well as in PD patient-derived iPSCs (Celardo et al. 2016; Gautier et al. 2016; Valadas et al. 2018), suggesting that dysfunctional interorganelle communication could contribute to pathogenesis. One question that arises is how parkin mutations affect other contacts, mediated by DJ-1 and the Grp75-VDAC1-IP3R complex, and how perturbation of one tethering complex affects the contacts mediated by others, again highlighting the need for an integrated network approach to interorganelle communication.
Parkin also modulates contacts in the endolysosomal pathway. Ubiquitination of Rab7 K38 by parkin increases Rab7 protein level and promotes its affinity for its downstream effector RILP, inducing dynein/dynactin-dependent transport toward the minus-end of microtubules (Song et al. 2016). Upon sensing cholesterol in late endosomes, the lipid transfer protein ORP1L forms contact sites between the late endosome and ER by connecting the Rab7-RILP complex with VAPA/B. This enables recruitment of the p150Glued subunit of dynactin, and subsequent transport of late endosomes to the center of the cell (Rocha et al. 2009). As we described above, interaction of VAPA/B and Rab7 with protrudin and FYCO1 mediates late endosome transport in the opposite direction, suggesting cholesterol and amino acids levels in late endosomes determine which complex gets formed and therefore the direction of endosome transport in response to the metabolic state of the cell. A significant decrease in Rab7 levels, as well as perturbations in downstream lysosomal morphology, were detected in parkin-deficient fibroblasts, suggesting perturbed ER-endosome contacts. The Rab7-RILP-ORP1L interaction also regulates autophagosome dynamics. When sufficient cholesterol is present, this complex can promote autophagosome transport and fusion with late endosomes. However, when cholesterol levels are low, ORP1L instead links Rab7/RILP to VAPA, forming ER-autophagosomal contacts (Wijdeven et al. 2016). It is an open question as to whether and how parkin regulates these contacts. Interestingly, CLN1 mutations, which cause a form of neuronal ceroid lipofuscinosis similar to CLN8, also suppress Rab7-RILP interaction, leading to impaired autophagy (Sarkar et al. 2020).
Leucine-rich repeat kinase 2 (LRRK2), a commonly mutated gene in familial PD, acts upstream of parkin in regulating MERC stability (Toyofuku et al. 2020). In its inactive form, LRRK2 interacts with parkin and the related E3 ubiquitin kinases MARCH5 and MULAN, blocking their activation by PERK under conditions of ER stress. In the same study, Toyofuku et al. (2020) show that a PD-associated LRRK2 mutation (LRRK2 G2019S) that enhances its kinase activity binds less parkin, permitting parkin activation. As a result, the levels of a subset of MERC proteins including Mfn1, Mfn2, and PTPIP51 are reduced and MERC abundance decreases. Mitochondrial fragmentation is increased in LRRK2 G2019S cells, and markers of mitochondrial activity, including citrate synthase activity, oxygen consumption rate, ATP synthesis, and Ca2+ buffering capacity, all decrease. Whether these results will hold true in patients with LRRK2 mutations is yet to be seen.
α-Synuclein, another PD-associated gene, has also been linked to MERCs. While data supporting the localization of α-synuclein at MERCs are somewhat limited (Calì et al. 2012; Ottolini et al. 2013), functionally it appears to regulate MERC stability. α-Synuclein binds VAPB, and overexpression of either wild-type or PD-associated mutant α-synuclein disrupts the VAPB-PTPIP51 interaction. This ultimately reduces ER-mitochondria contacts, disrupts Ca2+ exchange, and decreases mitochondrial ATP production (Paillusson et al. 2017). Overexpression of VAPB suppresses the effects of α-synuclein on MERCs and Ca2+ homeostasis. Despite a number of links between PD-associated genes and contact sites, the contribution of their disruption to disease pathogenesis has not been fully explored.
The expansive functional relevance of interorganelle communication raises the need to evaluate distinct contact sites not only in age-related pathologies, but also in physiological aging. Physiological aging results in accumulating damage to various macromolecules and the progressive decline of organelle function, and many of the interorganelle communication-linked processes mentioned in this review, like degradation, ROS production, and autophagy, are considered hallmarks of aging. Organelle morphology also changes with age (Monteiro 1991). For example, vacuoles increase in size, and while the cell’s volume expands as well, the ratio of vacuolar to cellular size increases over time. Age-dependent increased mitochondrial fragmentation is well established. While it is likely to expect that these changes in organelle morphology may be associated with perturbed contact sites, whether and how interorganelle communication is linked with physiological aging has still not been directly investigated. Does interorganelle communication differ between stem cells versus senescent cells, young versus aged cells, cells from increased health span mutants versus reduced longevity mutants? Does protein and/or lipid composition at contacts change with age? Do physiological functions at contact sites change, even in the absence of apparent morphological changes? If distinct contact sites do indeed change with age, can we identify the specific changes that contribute to aging or senescence? Are different cells or tissues more susceptible to changes in interorganelle communication, and could this underlie their selective vulnerability in aging and age-related pathologies? Recently, contacts between the ER and mitochondria were artificially increased by introducing a synthetic tether in Drosophila, which caused fragmentation of the mitochondrial network and increased production of ROS (Garrido-Maraver et al. 2020).While both are features normally associated with progressive aging and senescence, this study in flies has reported increased longevity and enhanced locomotion, raising further questions as to how manipulating contact sites could affect aging.
A systems approach to tackling cellular aging and neurodegeneration
In light of the work on contact sites, a redefined view of the cell emerges in which organelles and their associated processes do not function in isolation, but within a robust cellular communication network—the cellular communicome—to maintain homeostasis (Fig. 1). The concept of a cellular communicome represents the interconnectivity and interdependence of cellular organelles and the functions they regulate. Here, we highlighted novel findings regarding the cellular communicome and how its collapse could contribute to aging and neurodegeneration (Fig. 6). As our knowledge of contact sites expands, we find that many proteins participate at multiple distinct contact sites between different pairs of organelles, although the interconnectivity and balance between these contact sites is still predominantly unknown. For example, VAPA/B are a great example of proteins participating at a multitude of distinct sites. Likewise, contacts between organelles can be mediated by a number of distinct protein interactions, but whether they facilitate unique functions or are in part redundant is not fully understood. The many proteins that mediate MERCs exemplify this. The majority of studies so far have examined only one singular type of contact site with few associated functions. However, a given organelle probably simultaneously engages in large numbers of distinct interorganellar contacts at different locations in the cytoplasm at different times in response to various physiological stimuli, directing future studies to view distinct contacts and the proteins that mediate them as part of an integrated cellular network. This also holds true when trying to identify relevant defects involved in a given disease. A major challenge in studying cellular biology of disease is to tease apart causes from downstream secondary consequence of the disease. We feel that changing from a reductionist view to a more integrated systems approach holds great promise in addressing these important questions.
Advances in various technologies (Scorrano et al. 2019; Huang et al. 2020) are essential to be able to visualize, identify, and functionally evaluate the dynamic interplay between distinct contact sites and provide greater understanding of the cellular communicome. Electron microscopy approaches like FIB-SEM (Xu et al. 2019), and automated reconstruction with technologies like deep learning (Liu et al. 2020), are enabling direct visualization of cellular organelles and providing an in-depth view of contact sites. Applying such techniques to cells from patients or cells with specific engineered disease mutations could enable unbiased global analysis of perturbed contact sites in various pathologies, identifying the potentially relevant sites. Cryo-EM tomography can inform us on the molecular architecture of such distinct contact sites (Collado et al. 2019). Split fluorophores and multiplex proximity ligation in situ allow visualization of distinct contact sites that is compatible with genetic and CRISPR screens, where systematic analysis of disease-causing mutations can be performed. Spectral imaging (Valm et al. 2017) and reversible dimerization-dependent fluorescent proteins (Lee et al. 2020) tagged to contact site proteins can provide a dynamic view of cellular contacts of interest. Proximity biotinylation and split-proximity biotinylation approaches (Schopp et al. 2017; Cho et al. 2020) combined with proteomics can enable precise identification of protein components at distinct contact sites and how that composition changes with a particular disease mutation or age. Combining some of these visualization approaches with acute perturbations of various physiological stimuli could enable us to map out the landscape of interorganelle communication in healthy cells and tease apart relevant perturbations leading to disease. Furthermore, it seems quite likely that more contact sites and contact site proteins will be associated with aging and age-related pathologies, underscoring the need for more research to identify the entire repertoire of interorganelle communication (Shai et al. 2018).
Considering the wealth of data and the complexity of organelle interactions, ideas from network and complex adaptive systems theory could help to improve our understanding of the cellular communicome (Gottschling and Nyström 2017). Complex adaptive networks all share similar characteristics that hold true for a cell: They are complex, adaptive, constantly interacting with their environment, capable of feedback, maintaining memory of previous events, and, most importantly, showing emergence, where network behavior is more than just the sum of the individual parts. Applying a complex adaptive systems approach, one might start asking on the network level what happens when you start taking out components of the network and how the system responds. What are the hot spots in the network? How many components of the network can you take out before the system breaks down? Can you boost the resiliency of the network to better withstand perturbation?
To be able to take a systems approach, we envision a shared open access resource that would integrate all these data in a virtual cellular communicome. While such efforts are too large of an undertaking for any individual laboratory, it could be achieved if aided by institutions like The Allen Institute for Cell Science or Howard Hughes Medical Institute Janelia. As our knowledge of the cellular communicome is expanding, computer modeling and systems approaches could guide future experiments and further refine our understanding, as well as identify targets for therapeutic development. As highlighted throughout this review, a repercussion of an interconnected cellular communication network is that disturbances can cause cascading effects throughout the cell. This may affect various processes, where perturbation may be compensated for on the level of the cell (concept of resiliency) or could lead to progressive failure of other interconnected systems (concept of domino effect). These observations also raise the possibility that if the treatment of the original insult is not applied early enough, it is possible that reverting the original insult may not suffice to stop progressive deterioration of the network. A holistic comprehension of the cellular communicome and associated dysfunction might provide therapeutic targets that are closer to the root of the problem or could bypass or compensate for the afflicted process. It is exciting to speculate that chemical and biological modulators of organelle contact sites could be the novel druggable targets for development of therapeutics (Magalhães Rebelo et al. 2020).