
Impact of Two Neuronal Sigma-1 Receptor Modulators, PRE084 and DMT, on Neurogenesis and Neuroinflammation in an Aβ1–42-Injected, Wild-Type Mouse Model of AD
By Emőke Borbély, Viktória Varga, Titanilla Szögi, Ildikó Schuster, Zsolt Bozsó, Botond Penke, and Lívia Fülöp
Excerpt of the article published in International Journal of Molecular Sciences 23, no. 5: 2514., 24 February 2022, DOI: https://doi.org/10.3390/ijms23052514
Editor’s Highlights
- In early Alzheimer’s disease (AD), neurogenic impairment can be observed, accompanied by hyperreactive astrogliosis.
- Neuronal sigma-1 receptors (S1Rs) ligands are recognized as promising therapeutic agents that may alleviate symptom severity of AD, possibly via preventing amyloid-β-(Aβ-) induced neurotoxicity on the endoplasmic reticulum stress-associated pathways.
- Highly selective S1R agonists, improved the proliferation and differentiation of hippocampal stem cells, manifesting in a quantitative increase in progenitor cells and immature neurons.
Abstract
Alzheimer’s disease (AD) is the most common form of dementia characterized by cognitive dysfunctions. Pharmacological interventions to slow the progression of AD are intensively studied. A potential direction targets neuronal sigma-1 receptors (S1Rs). S1R ligands are recognized as promising therapeutic agents that may alleviate symptom severity of AD, possibly via preventing amyloid-β-(Aβ-) induced neurotoxicity on the endoplasmic reticulum stress-associated pathways. Furthermore, S1Rs may also modulate adult neurogenesis, and the impairment of this process is reported to be associated with AD. We aimed to investigate the effects of two S1R agonists, dimethyltryptamine (DMT) and PRE084, in an Aβ-induced in vivo mouse model characterizing neurogenic and anti-neuroinflammatory symptoms of AD, and the modulatory effects of S1R agonists were analyzed by immunohistochemical methods and western blotting. DMT, binding moderately to S1R but with high affinity to 5-HT receptors, negatively influenced neurogenesis, possibly as a result of activating both receptors differently. In contrast, the highly selective S1R agonist PRE084 stimulated hippocampal cell proliferation and differentiation. Regarding neuroinflammation, DMT and PRE084 significantly reduced Aβ1–42-induced astrogliosis, but neither had remarkable effects on microglial activation. In summary, the highly selective S1R agonist PRE084 may be a promising therapeutic agent for AD. Further studies are required to clarify the multifaceted neurogenic and anti-neuroinflammatory roles of these agonists.
1. Introduction
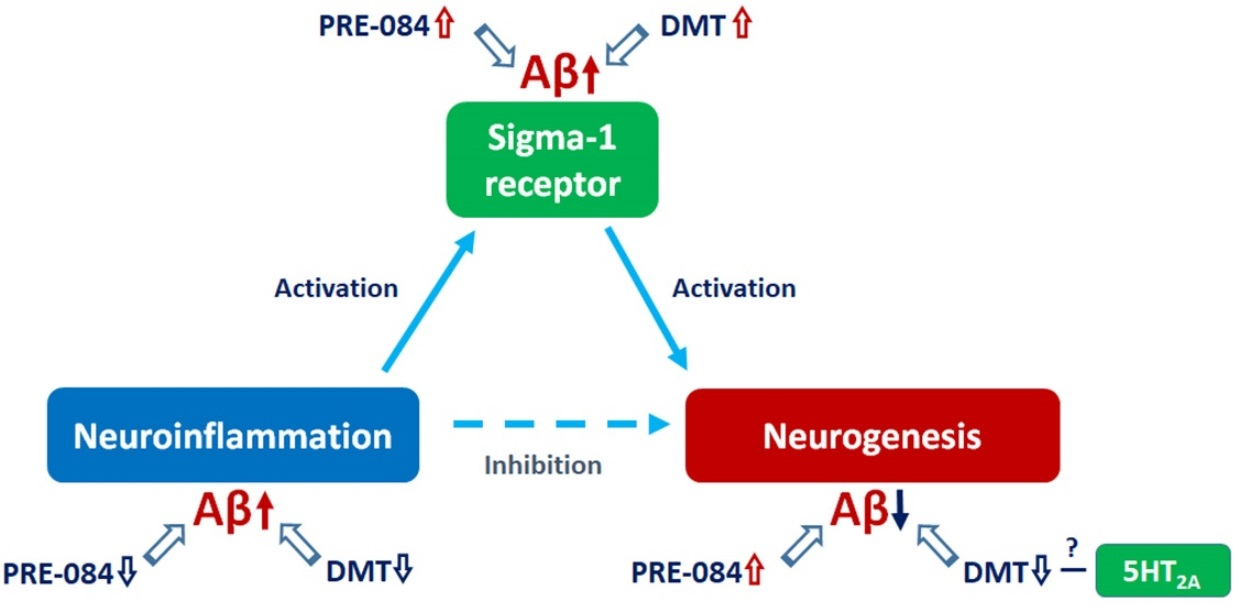
Alzheimer’s disease (AD) is the most common form of dementia, characterized by progressive memory loss, impaired learning, and cognitive dysfunction. The main pathological hallmarks of AD are extracellular amyloid plaques and intracellular neurofibrillary tangles accumulated in the cerebral tissue [1], which first appear in the hippocampal and entorhinal regions of the brain, explaining the impairment of cognitive functions [2]. These changes are accompanied by the damage of synaptic connections, and neuronal death. The abnormal cleavage of amyloid precursor protein (APP) by β- and γ-secretases predominantly yields 40 to 43 amino acid long amyloid-β (Aβ) peptides, which aggregate, and manifest as cerebral deposits. Besides forming plaques, these oligomeric forms of Aβ are also thought to be neurotoxic [3,4,5,6]. These short oligomers might interfere with crucial intracellular mechanisms and signaling pathways. Thus, they may affect cell homeostasis, proliferation, differentiation, and survival [7,8,9,10]. Another significant symptom of AD is neuroinflammation, which involves various inflammatory components, such as immune cells, cytokines, and chemokines. Neuroinflammation might significantly alter neurogenesis, as well as enhancing Aβ production and plaque formation [11,12,13]. Currently, there is no cure for AD, and its progression cannot be prevented; at present, only symptomatic treatments of mild to moderate efficiency are available. Therefore, effective disease-modifying therapeutics that may halt the progression of AD and contribute to the protection of neuronal integrity are eagerly awaited. A potentially new direction of the research aiming to find novel disease-modulating agents targets the sigma receptors (SRs). SRs have received considerable attention for their potential role in the prevention of Aβ-induced neurotoxicity, as well as in the regulation of the pathophysiology of AD. Furthermore, SRs may be essential for modulating neurogenesis in adulthood, and the stimulation of this process has been linked to AD. Thus, SR ligands are being recognized as promising therapeutic agents for treating or alleviating AD [6,14,15,16].
Two subtypes of SRs are distinguished, sigma-1 receptor (S1R) and sigma-2 receptor [17,18,19]. S1R is broadly expressed in the central nervous system (CNS), especially in the dentate gyrus (DG) region of the hippocampus (HC), both in neurons and glial cells. S1Rs are mainly located in a specific part of the cell where the endoplasmic reticulum (ER) and the mitochondria establish a tight interplay; this area is called the mitochondria-associated ER membrane (MAM) [16,20,21,22,23]. S1R is known to influence neuronal survival, proliferation, neurite growth, plasticity, as well as learning and memory functions [24,25,26,27]. It has been reported that the expression level of S1R decreases in patients with neurodegenerative diseases like AD [16,22,23,28,29,30,31,32,33].
S1R binds a diverse set of molecules, for example, antipsychotics, antidepressants, and neurosteroids [34,35,36,37]. A non-specific endogenous ligand of S1R is N,N-dimethyltryptamine (DMT), a hallucinogenic agent assumed to be produced in small quantities and accumulated in the CNS [16,38,39,40]. Previous studies have shown that the administration of DMT modulates many ion channels [39], protects against hypoxia-induced damage [41], alleviates neuroinflammation [42,43], increases the density of dendritic spines [44], as well as promotes neurogenesis and neuritogenesis [45,46,47,48,49]. However, DMT might also exert anxiogenic, neuro- and cytotoxic effects [47,50,51,52]. DMT is known to bind to several receptors with different affinities: 5-hydroxytryptamine (5-HT)1A-B, 5-HT1D, 5-HT2A-C, 5-HT5A, 5-HT6, 5-HT7 receptors, S1R, SERT, dopamine (D)1-5 receptors, α1AR, I1-3, TAAR, NMDA [53,54,55]. Several adverse effects of DMT are primarily associated with the stimulation of 5-HT2A receptors [47,50,51,53,56], while its positive impacts are rather related to the activation of S1Rs [40,41,42,43,44,46,49,50,52,57]. Moreover, the inflammation regulatory and plasticity promoting activities of DMT are also considered to result from its binding to both the S1Rs and 5-HT receptors. Identifying the valid contributor molecules and signaling pathways behind this assumption requires more convincing evidence.
Many exogenous ligands of S1R have been identified, including (+)-pentazocine, fluvoxamine, ANAVEX2-73, and 2-(4-morpholinethyl)-1-phenylcyclohexanecarboxylate (PRE084) [16,22,24,58]. The antidepressant and nootropic properties of PRE084 are also recognized [59]. Based on our current knowledge, PRE084 may promote neuroprotection and neurite growth by stimulating the expression of different neurotrophic factors, as well as by activating signaling pathways involved in cell survival [60,61,62,63,64,65]. Previous studies suggest that this S1R-agonist might positively impact learning and memory, as demonstrated in animal models of neurodegenerative diseases or traumatic brain injuries [63,64,66]. It is also reported that after the administration of Aβ25–35 infusion into the right lateral ventricle of mice, PRE084 administration has moderated the adverse behavioral effects of Aβ25–35 [27] via reducing neurotoxicity-induced cell death [32,64]. Moreover, PRE084 may also promote neurogenesis [9] and cell survival by attenuating excitotoxicity and reducing microglial activity, as well as diminishing the expression of proinflammatory factors [67,68].
As mentioned above, in addition to its ability to support cell survival under stress conditions, activated S1Rs may also stimulate the formation of new neurons, even in the adult brain. In adulthood, mammalian neurogenesis is derived from neuronal stem cells (NSCs) located in the subgranular zone (SGZ) of the dentate gyrus (DG) in the hippocampus (HC), as well as from NSCs in the subventricular region of the lateral ventricles [69,70]. After differentiation and migration, these newly formed neurons can integrate into local neuronal circuits of the HC; thus, they might have a significant role in plasticity, cognitive functions, learning, and memory processes [71]. An optimal microenvironment is essential for the division, differentiation, migration, and maturation of NSCs. Physiologically, the activity of adult hippocampal neurogenesis decreases with aging, leading to a usually mild, age-associated cognitive decline. However, a growing body of evidence indicates that the extent of adult neurogenesis is sharply diminished in the early stages of AD, even before the appearance of senile plaques [72,73,74,75,76,77,78]. This finding raises the question of whether impaired neurogenesis may initiate and/or contribute to more severe cognitive deficits, thus mediating AD’s pathogenesis. Furthermore, these findings suggest that the stimulation of neurogenesis might serve as a therapeutic target in AD, with a potential to improve cognitive functions and promote neural adaptability, thereby it might prevent or even treat AD.
In this study, two main objectives were addressed. First, to induce early acute AD-like impairments in neurogenesis and generate neuroinflammation in adult wild-type C57BL/6 mice by the intracerebroventricular (ICV) administration of Aβ1–42 oligomers. In this experimental paradigm, we followed the administration protocol described by Li et al., who examined the effects of Aβ25–35 on the same processes [9]. They reported that Aβ25–35 stimulated the proliferation of neuronal progenitor cells, while enhancing the death of newly formed neurons and impaired neurite growth. Secondly, we attempted to restore the normal functioning of adult neurogenesis and reduce neuroinflammation by activating S1Rs with two different ligands, PRE084 and DMT. The intraperitoneally-(IP-)-injected compounds were tested in wild-type mice, either treated with Aβ1–42-oligomers or injected with vehicle (phosphate buffered saline (PBS)) as a control. Based on previously published articles on the beneficial effects of these S1R modulators, we expected to detect an obvious positive impact of the tested agents on the Aβ1–42-induced impairments in adult neurogenesis and neuroinflammation [41,42,43,49,52,57,60,61,62,63,64,79].
2. Results
2.1. Effects of PRE084 and DMT on Adult Neurogenesis in Aβ1–42 and Vehicle-Treated Mice
Aβ1–42 and DMT impair, while PRE084 promotes the survival of progenitor cells in DG.
Proliferating cells were labeled by three IP injections of 5-Bromo-2′-Deoxyuridine (BrdU) with a 6 h interval, which was administered 24 h after the stereotaxic surgery. BrdU is a synthetic thymidine analog, which incorporates into the DNA strand, and can be detected by specific antibodies. We counted BrdU+ cells 14 days after the surgery. According to our results, the quantity of BrdU+ stem cells in the SGZ of the DG significantly differed among the six groups (ANOVA: p ≤ 0.0001). Aβ1–42 infusion significantly reduced the number of progenitor cells compared to the respective control group (PBS-PBS vs. Aβ1–42-PBS p = 0.001). Interestingly, significantly more severe negative changes were detected in animals treated with DMT. In those co-treated with both Aβ1–42 and DMT, hardly any BrdU+ stem cells were detected in the SGZ (Aβ1–42-DMT vs. PBS-PBS p ≤ 0.0001, vs. Aβ1–42-PBS p = 0.005, vs. Aβ1–42-PRE084 p ≤ 0.0001; PBS-DMT vs. PBS-PBS p = 0.001, vs. PBS-PRE084 p ≤ 0.0001). PRE084 treatment increased the amount of BrdU+ cells; the difference between the Aβ1–42-infused groups was significant (Aβ1–42-PBS vs. Aβ1–42-PRE084 p ≤ 0.0001) (Figure 1).
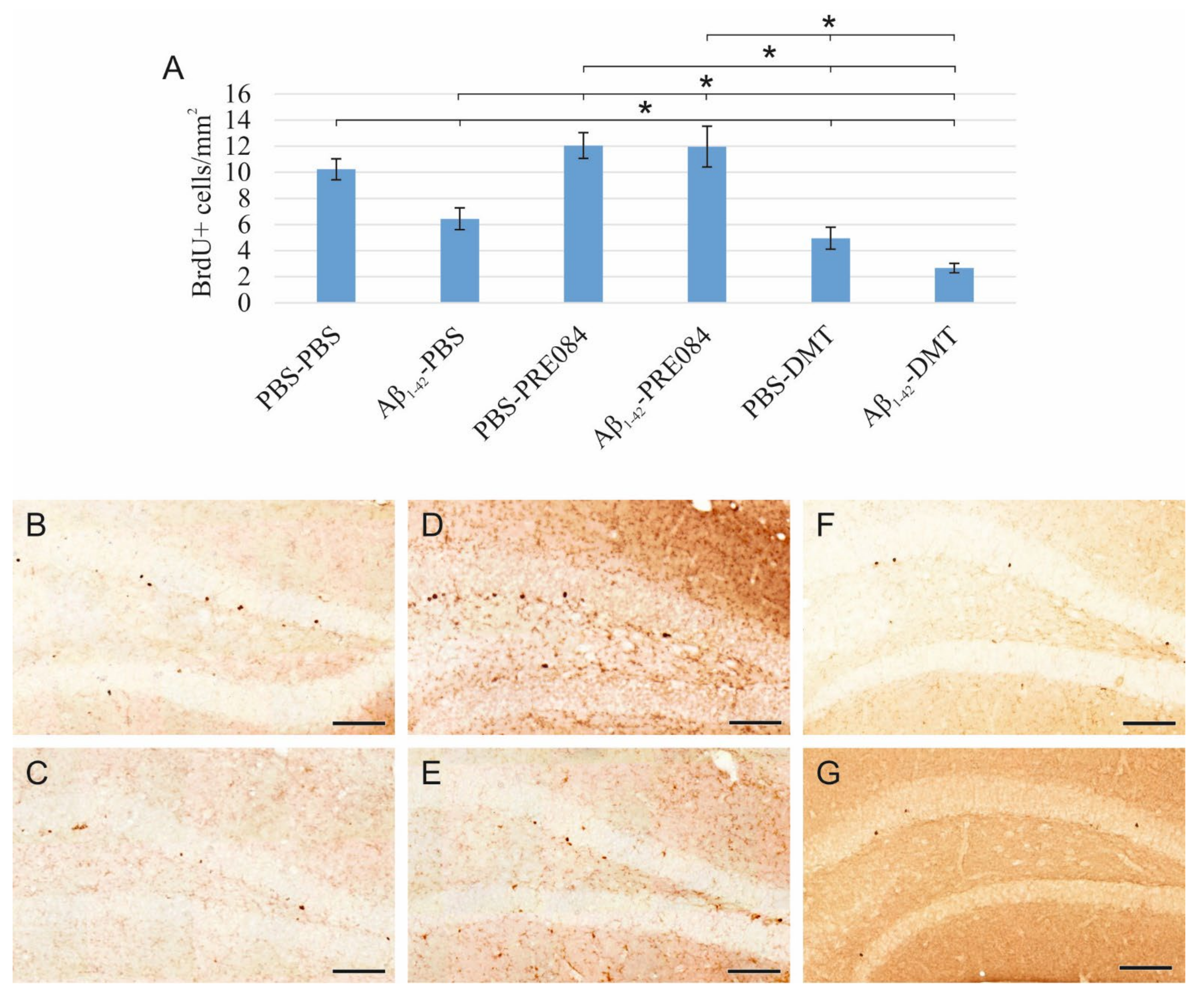
Results for 5-Bromo-2′-Deoxyuridine (BrdU) immunolabeling. We observed significant differences in the quantity of stem cells between the six groups (ANOVA: p ≤ 0.0001). Significantly fewer BrdU+ cells were detected in the Aβ1–42-PBS, PBS-DMT, and in the Aβ1–42-DMT treated animals compared to the PBS-PBS group (PBS-PBS vs. Aβ1–42-PBS p = 0.001, vs. PBS-DMT p = 0.001, vs. Aβ1–42-DMT p ≤ 0.0001). The difference between the Aβ1–42-PBS and Aβ1–42-DMT treatment groups was also significant (p = 0.005). PRE084-treatment increased the number of stem cells detected in the SGZ; this change was significant in the Aβ1–42-administered group compared to its vehicle-treated control (Aβ1–42-PBS vs. Aβ1–42-PRE084 p ≤ 0.0001). The differences between the following groups in pairwise comparisons also reached significance: PBS-PRE084 vs. Aβ1–42-PBS p ≤ 0.0001, vs. PBS-DMT p ≤ 0.0001, vs. Aβ1–42-DMT p ≤ 0.0001; Aβ1–42-PRE084 vs. PBS-DMT p ≤ 0.0001, vs. Aβ1–42-DMT p ≤ 0.0001. (B–G) Representative images of BrdU staining: (B) PBS-PBS, (C) Aβ1–42-PBS, (D) PBS-PRE084, (E) Aβ1–42-PRE084, (F) PBS-DMT, (G) Aβ1–42-DMT. Scale bars represent 100 μm. *: p ≤ 0.05.
Aβ1–42 and PRE084 increase the number of premature cells, while DMT does not affect their quantity.
To understand the effects of PRE084 and DMT on the maturation of granule cells, we quantified immature neurons in the SGZ of DG. To label premature cells, we stained a microtubule-associated protein called doublecortin (DCX), which is expressed specifically in migrating neuronal precursors. The measured DCX densities were significantly different among the six groups (ANOVA: p ≤ 0.0001). In those treated with Aβ1–42-PBS and PBS-PRE084, the number of immature neurons was significantly higher compared to the control group (PBS-PBS vs. Aβ1–42-PBS p = 0.037, vs. PBS-PRE084 p ≤ 0.0001, vs. Aβ1–42-PRE084 p ≤ 0.0001). We also detected a significant difference between the Aβ1–42-PBS and Aβ1–42-PRE084 mice groups (p = 0.007). DMT administration did not affect the number of premature neurons compared to PBS-PBS mice (Figure 2).
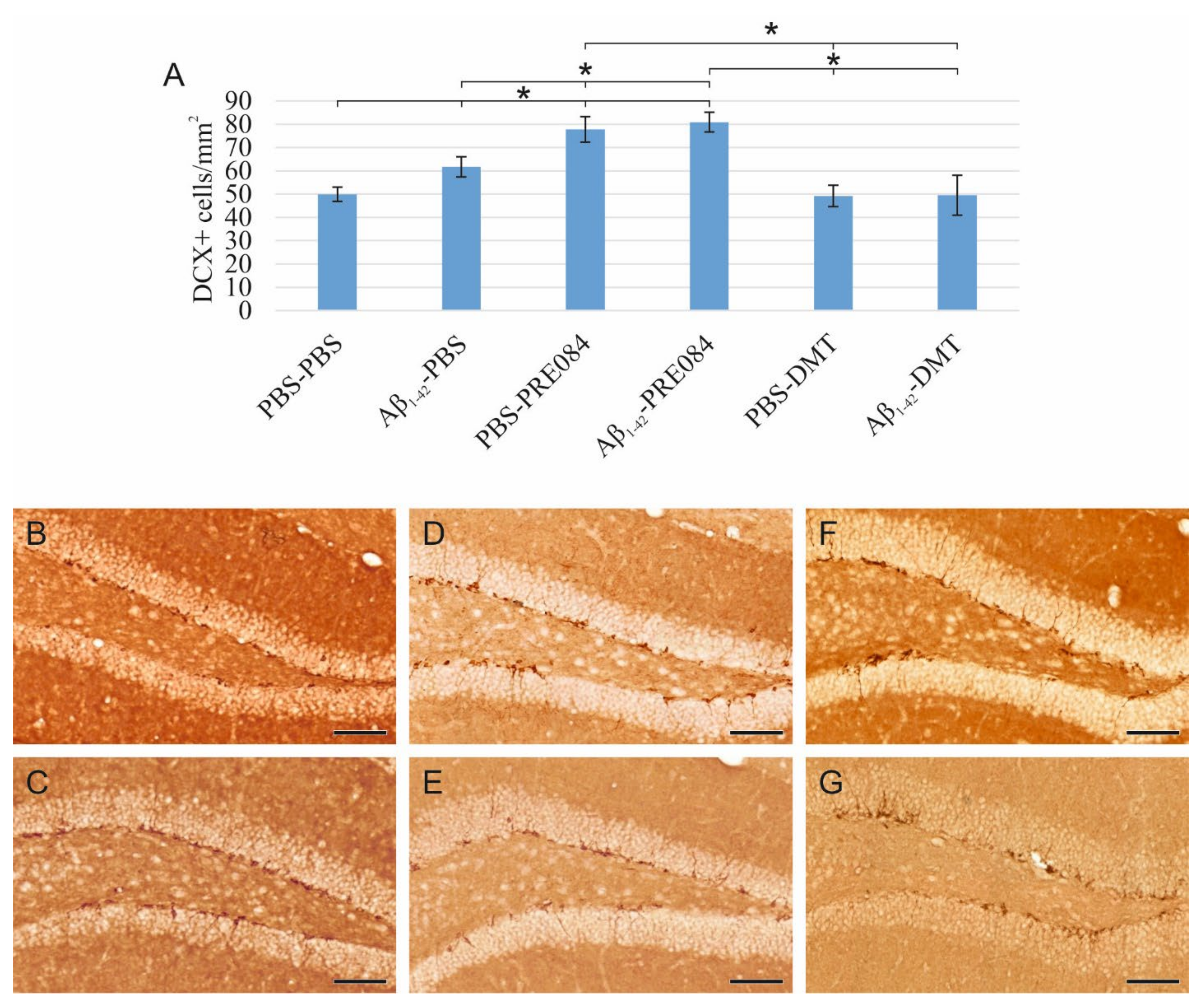
(A) Results for doublecortin (DCX) immunostaining. Detected DCX densities significantly differed among the six groups (ANOVA: p ≤ 0.0001). Compared to the control (PBS-PBS) animals, a significantly higher amount of DCX+ cells were detected in the Aβ1–42-PBS, PBS-PRE084 and Aβ1–42-PRE084-treated groups (PBS-PBS vs. Aβ1–42-PBS p = 0.037, vs. PBS-PRE084 p ≤ 0.0001, vs. Aβ1–42-PRE084 p ≤ 0.0001). Similarly, a significant difference was detected between the groups treated with Aβ1–42-PBS and Aβ1–42-PRE084 (p = 0.007). DMT treatment did not alter the number of immature neurons in the SGZ. Furthermore, significant differences were found when the groups were compared to the PBS-PRE084-treated group: PBS-PRE084 vs. Aβ1–42-PBS p = 0.023, vs. PBS-DMT p = 0.001, vs. Aβ1–42-DMT p = 0.001. Additional significant results were detected: Aβ1–42-PRE084 vs. PBS-DMT p ≤ 0.0001, vs. Aβ1–42-DMT p ≤ 0.0001. (B–G) Representative images of DCX immunolabeling: (B) PBS-PBS, (C) Aβ1–42-PBS, (D) PBS-PRE084, (E) Aβ1–42-PRE084, (F) PBS-DMT, (G) Aβ1–42-DMT. Scale bars represent 100 μm. *: p ≤ 0.05.
The density of mature granule cells is unaffected by Aβ1–42 or PRE084 administration, while DMT induces a decrease in neuronal density.
To detect and evaluate mature granule cells in the HC, we performed neuronal nuclei (NeuN) immunostaining (Figure 3). Again, significant differences were observed among the groups (ANOVA: p = 0.001). In DMT-treated animals, significantly lower NeuN+ cell densities were evident in the HC compared to the PBS-PBS and Aβ1–42-PBS group (PBS-PBS vs. PBS-DMT p = 0.001, vs. Aβ1–42-DMT p = 0.022; Aβ1–42-PBS vs. PBS-DMT p ≤ 0.0001, vs. Aβ1–42-DMT p = 0.003).
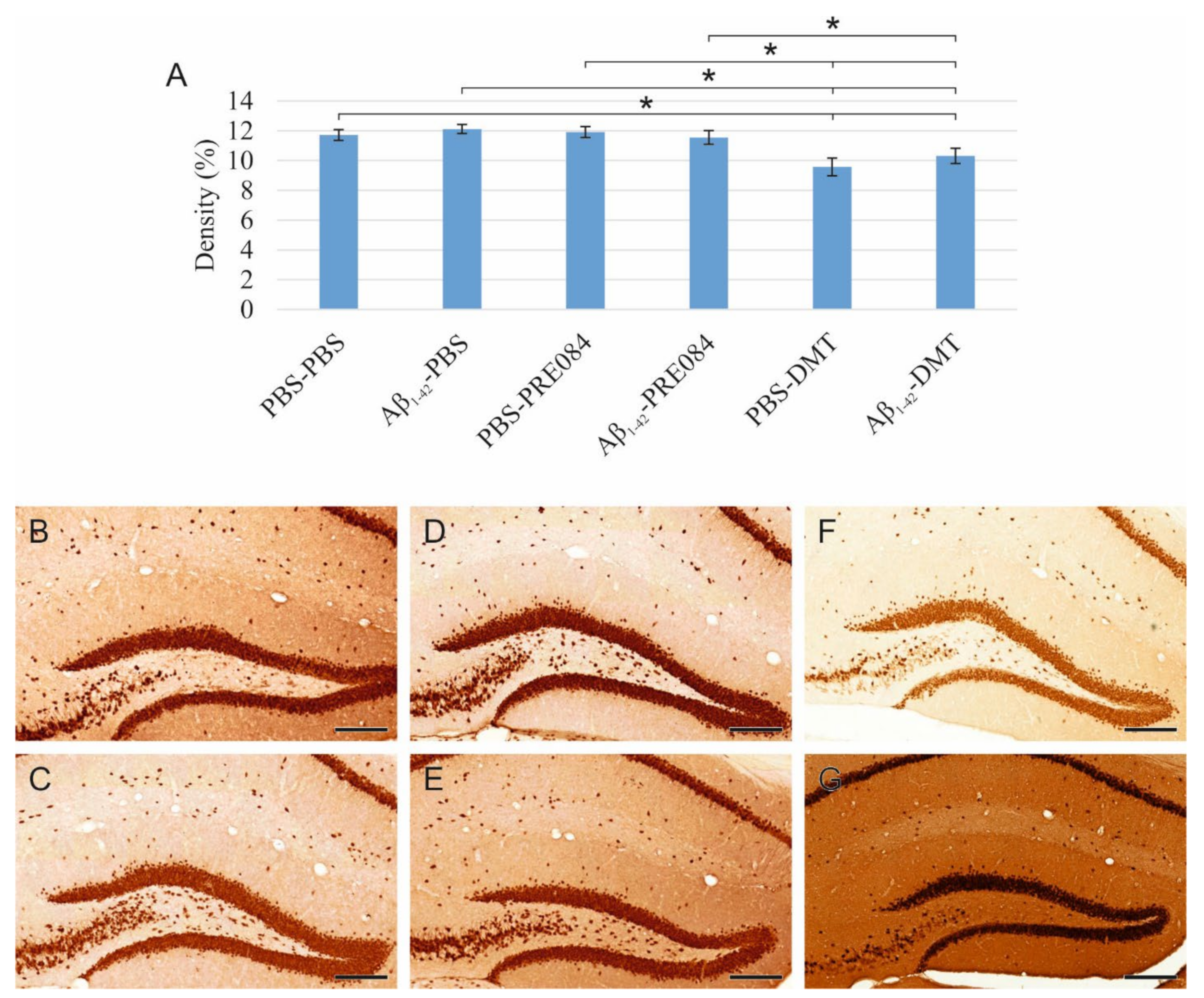
(A) Results for neuronal nuclei (NeuN) immunostaining. Significant differences were detected among the groups as follows (ANOVA: p = 0.001): in DMT-treated animals, significantly lower NeuN densities were evident compared to the PBS-PBS and Aβ1–42-PBS groups (PBS-DMT vs. PBS-PBS p = 0.001, vs. Aβ1–42-PBS p ≤ 0.0001; Aβ1–42-DMT vs. PBS-PBS p = 0.022, vs. Aβ1–42-PBS p = 0.003). Furthermore, significant differences were found when the groups were compared to the PBS-DMT-treated group: PBS-DMT vs. PBS-PRE084 p = 0.001, vs. Aβ1–42-PRE084 p = 0.006; Aβ1–42-DMT vs. PBS-PRE084 p = 0.024. (B–G) Representative photomicrographs of NeuN immunolabeling: (B) PBS-PBS, (C), Aβ1–42-PBS (D) PBS-PRE084, (E) Aβ1–42-PRE084, (F) PBS-DMT, (G) Aβ1–42-DMT. Scale bars represent 200 μm. *: p ≤ 0.05.
2.2. Effects of PRE084 and DMT on Neuroinflammation Induced by Aβ1–42
Aβ1–42 stimulates microglia activation, and neither PRE084, nor DMT alleviate this effect, while DMT alone significantly decreases microglial density.
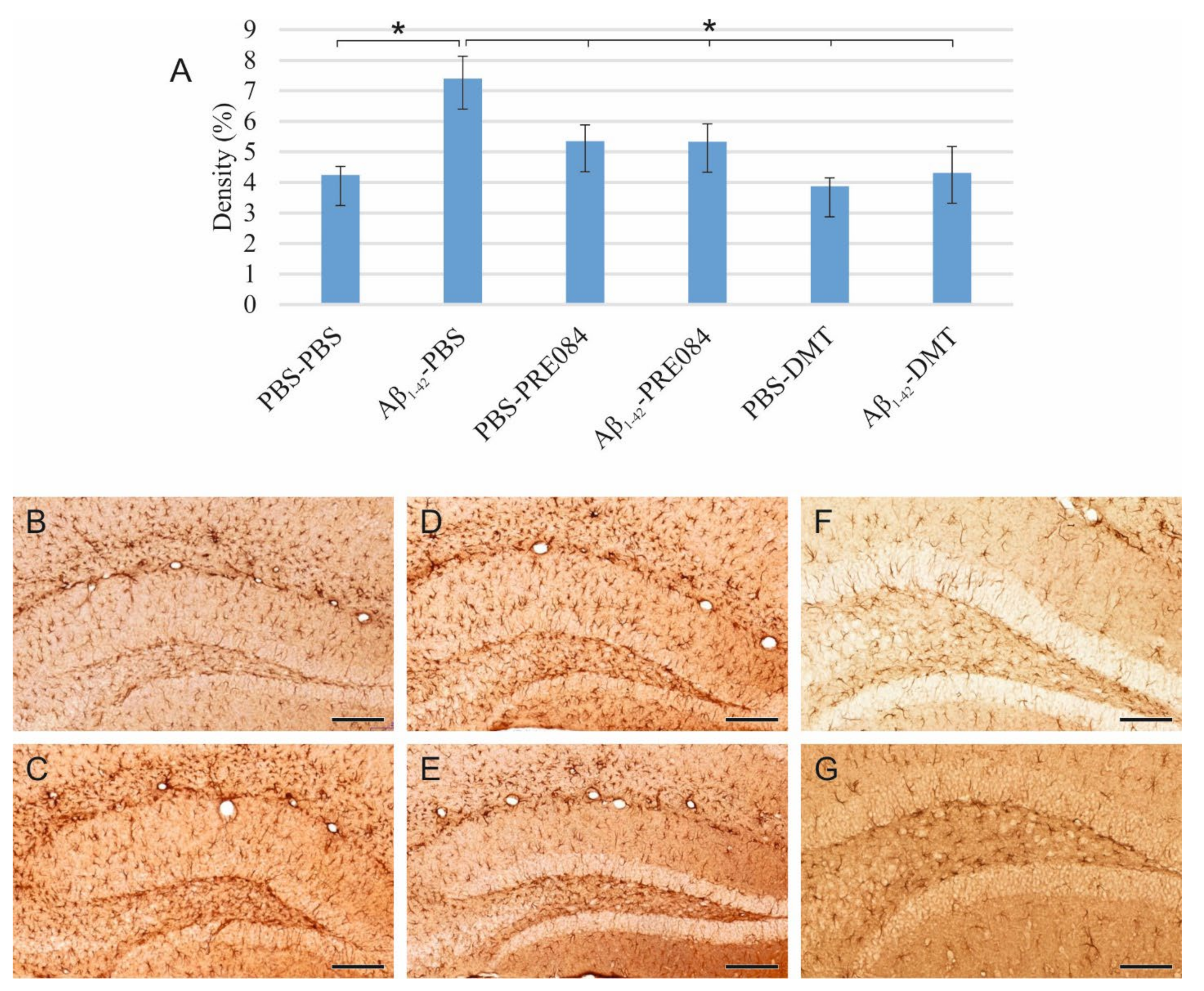
Neuroinflammation results from the activation of an immune response in the CNS, mediated by microglia and astrocytes. This process is induced by infective agents, neurodegenerative diseases, or injuries. To identify activated microglia in the HC, we stained ionized calcium-binding adapter molecule 1 (Iba1), expressed explicitly by monocyte-derived and resident macrophages, including microglia. Our results showed a significant difference in the density of Iba1+ microglia among the groups (ANOVA: p = 0.002). Aβ1–42 administration significantly increased the density of activated microglia compared to the vehicle-treated control groups (PBS-PBS vs. Aβ1–42-PBS p = 0.015; PBS-PRE084 vs. Aβ1–42-PRE084 p = 0.035; PBS-DMT vs. Aβ1–42-DMT p = 0.039). In the PBS-DMT group, the density of Iba1+ microglia was significantly reduced compared to PBS-PBS-treated animals (PBS-PBS vs. PBS-DMT p = 0.031). Still, none of the treatments were found to be able to alleviate the proinflammatory effect of Aβ1–42 (Figure 4).
(A) Results for ionized calcium-binding adapter molecule 1 (Iba1) immunolabeling. Significant differences were observed among the groups (ANOVA: p = 0.002). Aβ1–42 increased the density of Iba1+ microglia significantly compared to PBS-PBS, PBS-PRE084, and PBS-DMT treated mice, respectively (PBS-PBS vs. Aβ1–42-PBS p= 0.015; PBS-PRE084 vs. Aβ1–42-PRE084 p = 0.035; PBS-DMT vs. Aβ1–42-DMT p = 0.039). The difference between the PBS-PBS and PBS-DMT groups was also significant (PBS-PBS vs. PBS-DMT p = 0.031). Moreover, significant differences were detected between the following groups: Aβ1–42-PBS vs. PBS-PRE084 p = 0.005, vs. PBS-DMT p ≤ 0.0001; Aβ1–42-PRE084 vs. PBS-DMT p = 0.002. (B–G) Representative images of Iba1 immunostaining: (B) PBS-PBS, (C) Aβ1–42-PBS, (D) PBS-PRE084, (E) Aβ1–42-PRE084, (F) PBS-DMT, (G) Aβ1–42-DMT. Scale bars represent 100 μm. *: p ≤ 0.05.
Aβ1–42 stimulates astrocyte reactivation, while the administration of DMT or PRE084 reduces this effect.
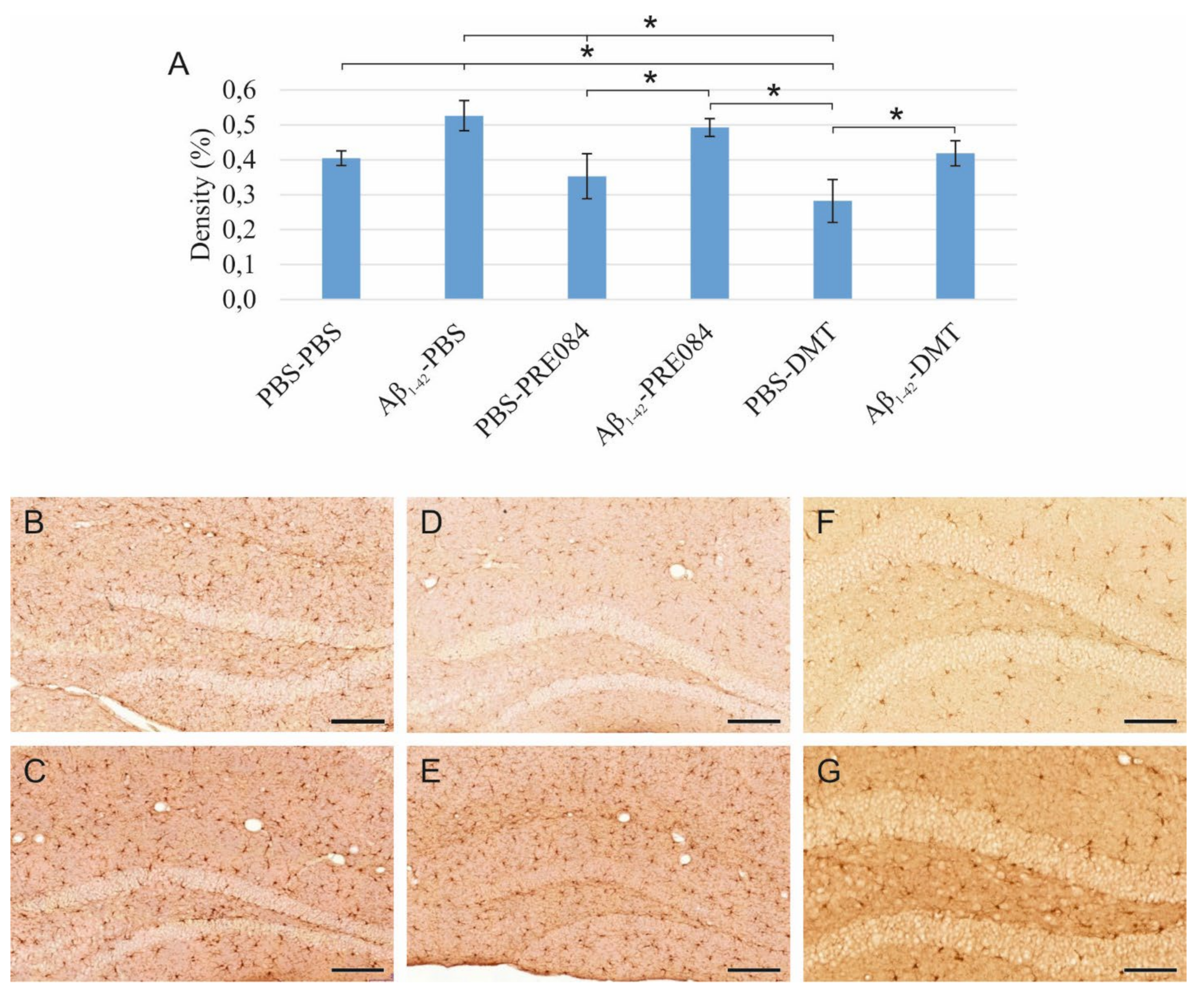
Reactive astrocytes were immunostained for glial fibrillary acidic protein (GFAP), an intermediate filament protein expressed by different cell types, mainly reactive astrocytes, in the CNS. Significantly different GFAP+ cell densities were detected in the HC of the different groups (ANOVA: p = 0.002). A significant increase in the rate of reactivated astrocytes was detected in the Aβ1–42-PBS group compared to PBS-PBS-treated mice (p ≤ 0.0001). Furthermore, GFAP+ cell densities were significantly lower in all other groups compared to Aβ1–42-PBS-treated mice (Aβ1–42-PBS vs. PBS-PRE084 p = 0.013, vs. Aβ1–42-PRE084 p = 0.013, vs. PBS-DMT p ≤ 0.0001, vs. Aβ1–42-DMT, p = 0.001). The stimulatory effect of Aβ1–42 on astrocyte reactivation was alleviated by PRE084 and DMT administration (Figure 5).
(A) Results of glial fibrillary acidic protein (GFAP) immunostaining. The densities of GFAP+ astrocytes differed among the groups (ANOVA: p ≤ 0.0001). A significantly higher GFAP+ density was detected in the Aβ1–42-PBS group compared to those treated with PBS-PBS (p ≤ 0.0001), PBS-PRE084 (p = 0.013), Aβ1–42-PRE084 (p = 0.013), PBS-DMT (p ≤ 0.0001), and Aβ1–42-DMT (p = 0.001). (B–G) Representative images of GFAP immunolabeling: (B) PBS-PBS, (C) Aβ1–42-PBS, (D) PBS-PRE084, (E) Aβ1–42-PRE084, (F) PBS-DMT, (G) Aβ1–42-DMT. Scale bars represent 100 μm. *: p ≤ 0.05.
The activation of inflammatory processes was assessed by the determination of certain proinflammatory cytokines (IL1β and TNFα). The levels of both pro- IL1β and soluble IL1β, as well as membrane-bound TNFα and soluble TNFα, were determined by western blot analyses (see Supplement Figure S1). These results corroborate our findings regarding the activation of the glial immunodefense system in response to the Aβ1–42 stimulus. The production of the active cytokine forms could be modulated by DMT-treatment; however, only the change in TNFα-level was significant.
2.3. S1R Protein Level Is Elevated by Aβ1–42 Treatment, as Well as by the Co-Administration of Aβ1–42 and PRE084 or DMT
To determine the effects of Aβ1–42 and PRE084 or DMT on the expression of S1R, a western blot (WB) analysis using GAPDH loading control was performed on HC and cerebral cortex samples of three animals per group. Our findings revealed a significant difference in the S1R levels among the groups (ANOVA: p ≤ 0.0001). S1R protein levels were significantly elevated in all groups, except in PBS-DMT-treated animals, as compared to control subjects (PBS-PBS vs. Aβ1–42-PBS p ≤ 0.0001, vs. PBS-PRE084 p = 0.018, vs. Aβ1–42-PRE084 p ≤ 0.0001, vs. PBS-DMT p = 0.540; vs. Aβ1–42-DMT p ≤ 0.0001, respectively). In comparison with Aβ1–42-PBS-treated mice, the Aβ1–42-PRE084 (p = 0.004) and Aβ1–42-DMT (p = 0.673) groups showed higher protein levels, while significantly lower levels of S1R were detected in PBS-PRE084 (p = 0.032) and PBS-DMT (p = 0.001) treated mice. As expected, the co-administration of Aβ1–42 and either of the S1R agonists increased the S1R protein level compared to the respective control group (Aβ1–42-PRE084 vs. PBS-PRE084 p ≤ 0.0001; Aβ1–42-DMT vs. PBS-DMT p = 0.015). Notably, the expression of S1R was significantly increased in Aβ1–42-PRE084-treated animals compared to the Aβ1–42-DMT group (p ≤ 0.0001). (Figure 6).
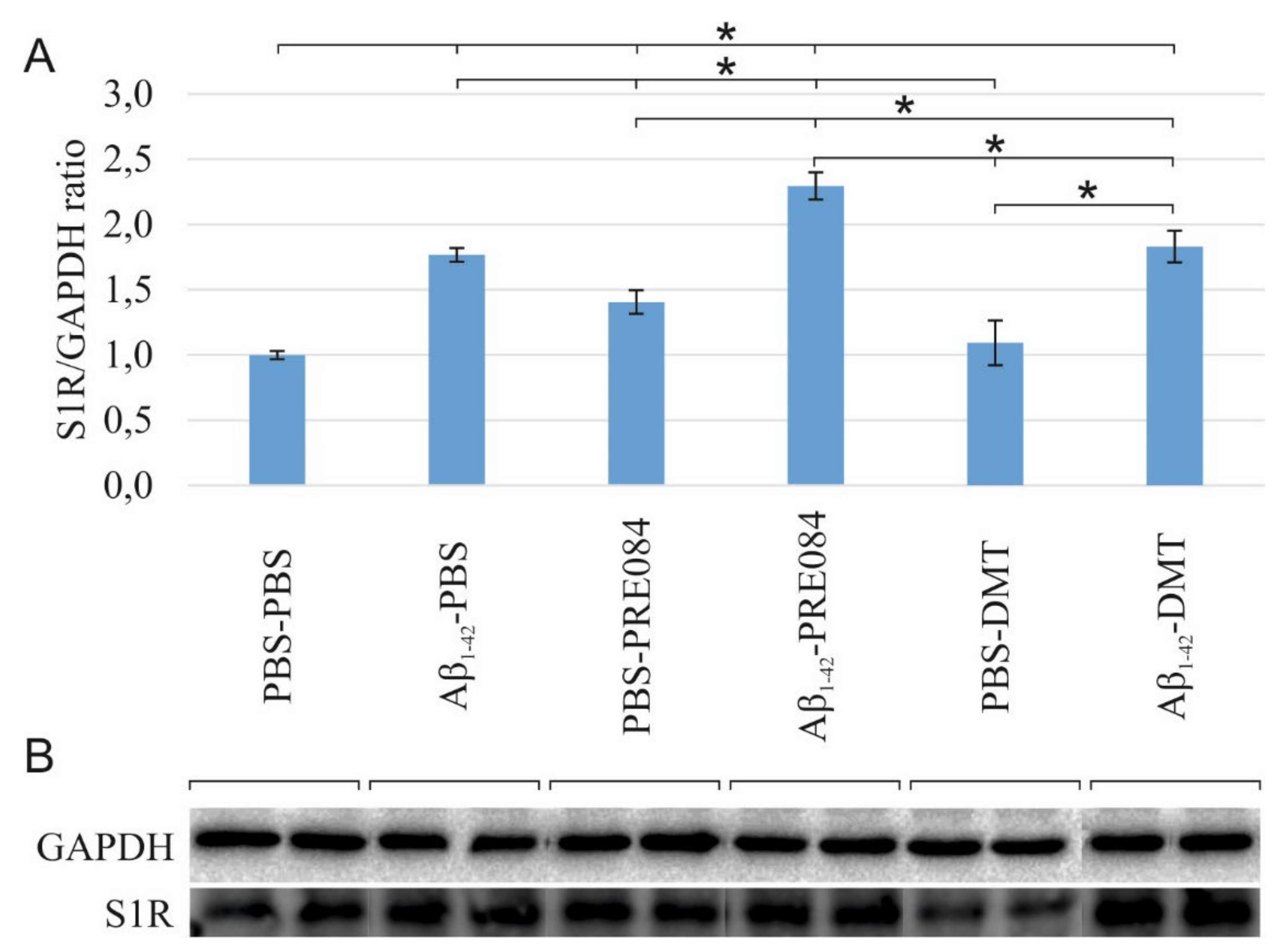
(A) Results for the western blot (WB) analysis. Significant differences were observed in the S1R levels among the groups (ANOVA: p ≤ 0.0001). Compared to PBS-PBS-treated mice, the S1R protein levels were significantly elevated in the Aβ1–42-PBS (p≤ 0.0001), PBS-PRE084 (p = 0.018), Aβ1–42-PRE084 (p ≤ 0.0001), and Aβ1–42-DMT (p ≤ 0.0001) groups. In PBS-DMT-treated mice, the S1R protein expression remained close to the control level (p = 0.540), while S1R levels were somewhat higher in the PRE084-treated groups (PBS-PBS vs. PBS-PRE084 p = 0.018; Aβ1–42-PBS vs. Aβ1–42-PRE084 p= 0.004; PBS-PRE084 vs. Aβ1–42-PRE084 p ≤ 0.0001). In contrast, the co-administration of Aβ1–42 and DMT induced a significant increase in the quantity of S1R (PBS-DMT vs. Aβ1–42-DMT p ≤ 0.0001). Furthermore, significant differences were detected in the S1R expression upon the pairwise comparisons of the following groups: Aβ1–42-PBS vs. PBS-PRE084 p = 0.032, vs. PBS-DMT p = 0.001; Aβ1–42-PRE084 vs. PBS-DMT p ≤ 0.0001, vs. Aβ1–42-DMT p ≤ 0.0001, respectively. (B) WB gel electrophoresis images of S1R and GAPDH lines of the experimental groups. *: p ≤ 0.05.
3. Discussion
During neurogenesis in adulthood, new neurons continuously develop and differentiate from hippocampal stem cells, and are integrated into existing neuronal networks to maintain plasticity of the CNS, and thereby preserve learning and memory functions. It has been recognized that the formation of new neurons reduces with age, manifesting in impaired cognitive functions [80]. In certain neurodegenerative diseases this cluster of mental symptoms is much more pronounced due to a decreased rate of neurogenesis, increased destruction of mature neurons, and enhanced neuroinflammatory responses. The most prevalent disease of this kind is AD, characterized by progressive dementia. Early alternations in adult neurogenesis and neuroinflammation may appear several years or even a decade before the diagnosis of AD, and probably contributes to the onset of neurological symptoms. It is hypothesized that an intensive stimulation of hippocampal neurogenesis and the reduction in neuroinflammation in adulthood could slow down the rate of decline of cognitive skills. Moreover, the uniquely structured S1R protein, functioning as a ligand-operated chaperone, is known to play a major role in both neurogenesis and neuroinflammation. Thus, it is assumed that the activation of S1Rs may be a promising therapeutic strategy to stimulate adult neurogenesis and alleviate neuroinflammatory processes.
The first objective of our study was to model these early alternations appearing in AD. Our experimental paradigm was based on the work of Li et al., in a modified way: instead of Aβ25–35, we injected Aβ1–42 ICV to induce early AD-like changes [9]. The reason for this modification is that Aβ25–35 is a non-natural, truncated sequence, and although it is prone to aggregation, its kinetics for aggregation differ from that of the native Aβ1–42 peptide. Therefore, using this latter peptide should yield biologically more relevant findings [81]. In the work of Li et al., neurogenesis was assessed 14 and 28 days after the peptide injections, and significant differences were detected on day 28 in neurogenic markers compared to baseline (reduced proliferation and neurite growth, increased death of newly formed cells) [9]. In our experimental model, AD-like cerebral neurogenic and neuroinflammatory changes could be detected as early as two weeks after the administration of Aβ1–42. We demonstrated that a single administration of Aβ1–42, directly into the lateral ventricles, significantly impaired the proliferation and increased the number of immature cells in mice. The effects of Aβ on neurogenesis are highly controversial in the literature. Numerous reports indicate that Aβ significantly decreases the formation of new neurons, possibly by impairing their ability to divide, as well as by diminishing the survival of neuronal stem cells in DG [7,8,9,75,76,77,82]. However, some research groups have published that Aβ can induce the initial proliferation step of neuron formation in different transgenic mouse strains [9,78,83,84,85] or in cellular models of AD [86,87,88,89,90,91]. In our experiments, an increase in the number of differentiating immature neurons was observed in Aβ1–42-treated animals, which may be explained by a compensatory cerebral mechanism [77,92]. Specifically, this enhancement of neuronal cell differentiation may be a response to the disturbed homeostasis resulting from the decrease in the stem cell population, aiming to restore the balance within the CNS. As we expected, in our experimental model, no significant reduction was detected in the density of mature, functional neurons in HC two weeks after the administration of Aβ1–42, indicating that the existing neuronal system may remain unaffected. Regarding neuroinflammation, we found that a single administration of Aβ1–42 stimulated neuroinflammatory processes, causing a significant increase in the densities of activated microglia and hyperreactive astrocytes. In line with our observations, several in vivo experiments have demonstrated the neuroinflammation-inducing effects of Aβ fibrils and oligomers injected into the brain tissue in different experimental models [93,94,95]. This neuroinflammatory environment may affect adult neurogenesis either positively or negatively [11,12,96,97,98,99,100,101]. It is known that cytokines and chemokines produced by activated microglia and astrocytes play an important role in neuroinflammatory processes. Certain anti-(IL-4, IL-10) and proinflammatory (IL-6, TNF-α) factors substantially influence neurogenesis, e.g., they can diminish proliferation and cell survival, while they may also stimulate cell differentiation [13]. Thus, beyond its direct effects on immature neurons, Aβ1–42 may also affect neurogenesis by generating a relatively mild, but chronic neuroinflammatory environment. Further research is needed to clarify the relative contribution of these two processes (direct and indirect) to the final decline of adult neurogenesis in AD.
Since the S1R protein plays a major role in neurogenesis and neuroinflammation, and changes in S1R expression levels have not been studied in exogenous Aβ-induced AD models, we examined the expression levels of this protein. In our case, the expression of S1R increased after a single administration of Aβ1–42. This finding may contradict some literature data, which report on the down-regulation of S1R in the early stage of human AD [24]. In the reported cases, both the amount and the binding potential of S1R were found to be decreased, presumably as a consequence of hippocampal neuronal death [24,102,103,104,105]. In contrast, other studies indicate that AD-related ER-stress can lead to an up-regulation of S1R [16,29,106,107], which, serving as a chaperon, modulates the canonical unfolded protein response (UPR) pathways (PERK, IRE1a, ATF6) [16,108]. In our study, the observed elevation of the level of S1R may be a consequence of the cytotoxic effect of Aβ1–42, which induces ER stress, and thus activates the UPR pathways and upregulates S1R expression.
To date, the biological effects of DMT and PRE084 have not been studied in an Aβ-induced model of early AD with demonstrated changes in neurogenesis and S1R expression levels, as well as neuroinflammation. Therefore, we aimed to assess whether the modulation of S1Rs with selected ligands can restore Aβ1–42-induced alternations in adult neurogenesis and reduce neuroinflammation.
In our study, DMT significantly reduced the number of neuronal stem cells and densities of neurons. Similar to this finding, another tryptamine, psilocybin (4-phosphoryloxy-N, N-dimethyltryptamine) with a chemical structure close to that of DMT and a high binding affinity to 5-HT2A receptors (Kd = 6 nM), was also found to impair synaptic growth and neurogenesis (proliferation and neuronal survival) [109]. However, the neuroprotective and neurogenesis stimulating effects of DMT and its analog, 5-methoxy-DMT, exerted via S1Rs, were also described in in vitro cell cultures and in a wild-type rodent model [44,46,49,54]. In our study, DMT was administered at a concentration of 1 mg kg–1, thus it is supposed to have occupied both receptor types, so their mixed effects could have been observed. Comparison of the Kd values (DMT-S1R Kd = 14.75 μM, DMT-5-HT2A receptor Kd = 130 nM) indicates that DMT binds to the 5-HT2Areceptor with higher affinity than to S1R; thus, it is more likely to act on the 5-HT2A receptors than on S1R [39,53]. Therefore, we suppose that DMT exerted its negative effect on neurogenesis via the 5-HT2A receptors. The results of our WB analysis support this hypothesis, since the expression of the S1R protein was only slightly elevated after DMT treatment.
Regarding the relation of DMT and neuroinflammation, conflicting findings are published in the literature. Some of them support the theory that DMT can alleviate neuroinflammatory processes, thus it may reduce the density of reactive astrocytes [41,42,43,52,57]. This effect may be related to the ability of DMT to bind to S1R [41,42,43,52], but the serotonergic receptors may also have roles in this process [110]. Morales-Garcia et al. reported that DMT induces a significant increase in the density of GFAP+ astrocytes via the activation of S1Rs, but these researchers conclude that this elevated GFAP level promotes neurogenesis [49]. In our experiments, DMT treatment was found to exert a positive effect on activated microglia and hyperreactive astrocytes against the Aβ1–42-induced neurotoxicity, but it was not detected to promote neurogenesis.
These contradictory results may be explained by the application of different protocols (injection and doses of BrdU and DMT, different survival times). It is also known that although DMT can penetrate the blood-brain barrier, upon exogenous administration its concentration in the CNS is elevated for a relatively short time only (elimination half-life ~15 min [44]). Therefore, it is also possible that in our model, the concentration of DMT in the CNS after IP administration was not sufficient to exert its effects on S1R as Morales-Garcia reported [49]. Further experiments are required to elucidate the exact mode of action of DMT regarding neurogenesis and neuroinflammation.
To study the effect of an exogenous S1R agonist on neurogenesis and neuroinflammation, we applied PRE084 (Kd = 2.2 nM, [111]). Similarly, as Li et al. reported in an Aβ25–35-induced mouse model of AD, we have demonstrated that PRE084 promotes neurogenesis upon treatment with Aβ1–42, as it is indicated by the quantitative increase in stem cells and immature neurons after PRE084 administration. Furthermore, PRE084 per se activates cell proliferation, possibly by stimulating S1R.
Regarding neuroinflammation, the density of hyperreactive astrocytes and the degree of Aβ1–42-induced astrogliosis were reduced by the administration of PRE084. However, the substance neither per se, nor in combination with Aβ1–42 could impair microglial activation. It is known that in case of CNS tissue damage, activated microglia may behave either neurotoxic or neuroprotective, depending on their morphological and functional states. According to the literature, PRE084 can stimulate the proliferation of the anti-inflammatory type of microglia (M2), while it suppresses pro-inflammatory M1 microglia, thus it maintains the delicate balance between functional restorative and inflammatory glial phenotypes [62,112]. As we did not analyze the distribution and morphology of the microglia, we assume that the apparent ineffectiveness of PRE084 treatment on microglial activation may result from the above mentioned two mutual processes.
PRE084 binds to S1R with high affinity, either alone (compared to PBS and DMT controls) and when co-administered with Aβ1–42 (compared to Aβ1–42-PBS or Aβ1–42-DMT animals), and significantly induces the expression of this receptor protein. These results may confirm that PRE084 activates the S1R receptors effectively, so its neurogenic impact is more pronounced than that of DMT.
…
5. Conclusions
Adult neurogenesis is essential for CNS plasticity. In early AD, neurogenic impairment can be observed, accompanied by hyperreactive astrogliosis. During the treatment of AD, neurogenesis should be promoted, while neuroinflammation should be suppressed. S1R plays a role in both processes. In our experiments, we established a model of early AD induced by Aβ1–42, in which acute neuroinflammation, impaired neurogenesis and elevated S1R levels were detected. In this model, two S1R agonists were tested. DMT, binding moderately to S1R but with a high affinity to 5-HT receptors, negatively influenced neurogenesis in the Aβ1–42-induced rodent model, probably explained by its acting on the latter receptor class. In contrast, the highly selective S1R agonist, PRE084 improved the proliferation and differentiation of hippocampal stem cells, manifesting in a quantitative increase in progenitor cells and immature neurons. Further experiments are required to investigate the main molecular pathways targeted by DMT, through which it affects neurogenesis and the survival of mature neurons. Moreover, DMT and PRE084 were found to significantly reduce Aβ1–42-induced hyperreactive astrogliosis. However, none of these ligands had a remarkable effect on microglial activation. Therefore, further studies are needed to clarify the role of DMT and PRE084 in neuroinflammatory processes induced by Aβ1–42, resembling the changes characteristic of AD.