
High Soluble Amyloid-β42 Predicts Normal Cognition in Amyloid-Positive Individuals with Alzheimer’s Disease-Causing Mutations

By Andrea Sturchio , Alok K Dwivedi, Tarja Malm, Matthew J A Wood, Roberto Cilia, Jennifer S Sharma, Emily J Hill, Lon S Schneider, Neill R Graff-Radford, Hiroshi Mori, Georg Nübling, Samir El Andaloussi, Per Svenningsson, Kariem Ezzat, Alberto J Espay, and Dominantly Inherited Alzheimer Consortia (DIAN)
Extract of the article published in Journal of Alzheimer’s Disease, 2022 Sep 16. doi: 10.3233/JAD-220808. Epub ahead of print. PMID: 36120786.
Editor’s Highlights
- Mutations in any of three genes (APP, PSEN1, and PSEN2) lead to Alzheimer’s disease (AD) with complete penetrance.
- The genetic evidence causally implicates the fibrillogenic 42-amino acid amyloid-β peptide (Aβ42).
- While insoluble amyloid plaques can be present in normal individuals, low soluble levels of Aβ42 are an invariable feature of AD.
- Cognitive deterioration was strongly predicted by lower levels of soluble Aβ42 but not by increases in SUVR, CSF phosphorylated tau (p-tau) or total tau (t-tau)
- Brain toxicity in AD may be predominantly mediated by a reduction of the soluble protein pool, its functional fraction, rather than its accrual into amyloids.
Abstract
Background: In amyloid-positive individuals at risk for Alzheimer’s disease (AD), high soluble 42-amino acid amyloid-β (Aβ42) levels are associated with normal cognition. It is unknown if this relationship applies longitudinally in a genetic cohort.
Objective: To test the hypothesis that high Aβ42 preserves normal cognition in amyloid-positive individuals with Alzheimer’s disease (AD)-causing mutations (APP, PSEN1, or PSEN2) to a greater extent than lower levels of brain amyloid, cerebrospinal fluid (CSF) phosphorylated tau (p-tau), or total tau (t-tau).
Methods: Cognitive progression was defined as any increase in Clinical Dementia Rating (CDR = 0, normal cognition; 0.5, very mild dementia; 1, mild dementia) over 3 years. Amyloid-positivity was defined as a standard uptake value ratio (SUVR) ≥1.42 by Pittsburgh compound-B positron emission tomography (PiB-PET). We used modified Poisson regression models to estimate relative risk (RR), adjusted for age at onset, sex, education, APOE4 status, and duration of follow-up. The results were confirmed with multiple sensitivity analyses, including Cox regression.
Results: Of 232 mutation carriers, 108 were PiB-PET-positive at baseline, with 43 (39.8% ) meeting criteria for progression after 3.3±2.0 years. Soluble Aβ42 levels were higher among CDR non-progressors than CDR progressors. Higher Aβ42 predicted a lower risk of progression (adjusted RR, 0.36; 95% confidence interval [CI], 0.19-0.67; p = 0.002) better than lower SUVR (RR, 0.81; 95% CI, 0.68-0.96; p = 0.018). CSF Aβ42 levels predicting lower risk of progression increased with higher SUVR levels.
Conclusion: High CSF Aβ42 levels predict normal cognition in amyloid-positive individuals with AD-causing genetic mutations.
INTRODUCTION
Key support for the toxic amyloid hypothesis comes from the observation that mutations in any of three genes (APP, PSEN1, and PSEN2) lead to Alzheimer’s disease (AD) with complete penetrance [1]. The genetic evidence causally implicates the fibrillogenic 42-amino acid amyloid-β peptide (Aβ42). However, the disease pathogenesis may arise from either of two ends of the protein aggregation process: the increase in insoluble amyloid plaques or the depletion of the soluble Aβ42 peptide, which has important functions. While insoluble amyloid plaques can be present in normal individuals, low soluble levels of Aβ42 are an invariable feature of AD [2–4].
The hypothesis of Aβ toxicity has traditionally been supported by the notion that AD-causing mutation carriers must have high levels of soluble Aβ42 relative to non-mutation populations. In fact, mutation carriers have lower Aβ42 levels compared to non-mutation populations [3]. The reduction in soluble Aβ42 levels among mutation carriers begins as many as 25 years before the onset of cognitive symptoms [4]. Therefore, the toxicity in the process of accelerated protein aggregation among mutation carriers may conceivably be due to the depletion in soluble Aβ42 to a greater extent than the corresponding increase in amyloid [5]. This alternative hypothesis offers an explanation for the failures in translating amyloid reduction into cognitive improvement [6], even among mutation carriers [7], and for the paradoxes posed by the large proportion of amyloid-positive individuals without dementia and even of centenarians without history of cognitive abnormalities, half of whom have autopsy-confirmed AD pathology [8–10].
We recently observed that among amyloid positron emission tomography (PET)-positive individuals, higher levels of soluble Aβ42 were associated with normal cognition and brain volumes in all tertiles of brain amyloidosis, with an effect size greater than that of increases in brain amyloid burden [11]. We here tested the hypothesis that in amyloid PET-positive individuals with AD-causing APP, PSEN1, or PSEN2 mutations, higher CSF Aβ42 levels reduce the risk of cognitive progression to a greater extent than lower levels of brain amyloidosis and lower levels of CSF phosphorylated tau (p-tau) and total tau (t-tau).
RESULTS
Of 534 subjects participating in the DIAN study (mean age, 38±11.1 years), 232 mutation carriers met eligibility criteria (mean age, 38.3±11 years). Among them, 191 had available PiB-PET data at baseline (mean SUVR, 1.9±1.0), of whom 108 were PiB-PET-positive (mean SUVR, 2.5±1.0) (Table 1). These subjects were followed for a mean of 3.3±2.0 years (range = 1, 9). Because of missing data on some CSF and SUVR levels, multivariable analyses were based on 93 PiB-PET-positive samples, 85 PiB-PET-positive samples (SUVR ≥1.49), and 162 overall samples which include PiB-PET-positive and negative samples (Supplementary Figure 1). No differences in the baseline characteristics except CDR at baseline were observed between cohorts with missing versus without missing data (Supplementary Table 1).
| PiB-PET-positive cohort (n = 108) | Overall cohort (n = 232) | |
| Age (y) | 41.0 (10.4) | 38.3 (11.0) |
| Sex (female) | 55 (50.9%) | 130 (56.0%) |
| Education (y) | 13.8 (3.0) | 14.3 (3.0) |
| APOE4 carriers | 40 (37.0%) | 72 (31.0%) |
| CSF Aβ42 (pg/ml) | 264.6 (107.9)a | 350.6 (186.8)a |
| SUVR (amyloid-PiB-PET) | 2.5 (1.0)b | 1.9 (1.0)b |
| t-tau (pg/ml) | 144.5 (89.4)c1 | 111.2 (81.6)c1 |
| p-tau (pg/ml) | 81.7 (38.1)c2 | 61.9 (37.6)c2 |
| CDR at baseline | ||
| 0 | 55 (50.9%) | 147 (63.4%) |
| 0.5 | 33 (30.6%) | 60 (25.9%) |
| 1 | 17 (15.7%) | 20 (8.6%) |
| 2 | 2 (1.9%) | 3 (1.3%) |
| 3 | 1 (0.9%) | 2 (0.9%) |
| CDR progression | ||
| No | 65 (60.2%) | 165 (71.1%) |
| Yes | 43 (39.8%) | 67 (28.9%) |
| CDR (two consecutive visits) | ||
| <0.5 | 53 (49.1%) | 150 (64.7%) |
| ≥0.5 | 55 (50.9%) | 82 (35.3%) |
| CDR (any visit) | ||
| <1 | 74 (68.5%) | 176 (75.9%) |
| ≥1 | 34 (31.5%) | 56 (24.1%) |
| CDR-SB at baseline | 1.8 (2.8)d | 1.2 (2.5)d |
| CDR-SB at last visit | 3.7 (5.0)d | 2.7 (4.7)d |
| ≥4.5 | 74 (68.5%) | 178 (76.7%) |
| <4.5 | 34 (31.5%) | 54 (23.3%) |
| MMSE at baseline | 26.4 (4.9)e1 | 27.1 (4.4)e1 |
| MMSE at last visit | 23.4 (7.7)e2 | 25.1 (7.2)e2 |
| ≤24 | 69 (63.9%) | 174 (75.0%) |
| >24 | 39 (36.1%) | 58 (25.0%) |
| FDG-PET at baseline (SUVR) | 1.8 (0.3)f1 | 1.8 (0.2)f1 |
| FDG-PET last visit (SUVR) | 1.7 (0.3)f2 | 1.8 (0.3)f2 |
| Average hippocampi baseline (mm3) | 4022.4 (708.7)g1 | 4174.6 (626.0)g1 |
| Average hippocampi last visit | 3795.2 (767.6)g2 | 4039.4 (741.3)g2 |
| Normalized hippocampi baseline (mm3) | 4022.3 (709.6)g1 | 4174.3 (627.0)g1 |
| Normalized hippocampi last visit (mm3) | 3795.2 (767.6)g2 | 4039.4 (741.3)g2 |
Characteristics of study cohort
n, number of subjects; APOE4, Apolipoprotein ɛ4; CDR, clinical dementia rating; CDR-SB, CDR sum of boxes; CSF, cerebrospinal fluid; Aβ42, 42-amino acid amyloid-beta peptide; t-tau, total tau; p-tau, phosphorylated-tau; PiB-PET, Pittsburgh compound B positron emission tomography; SUVR, standardized uptake value ratio; MMSE, Mini-Mental State Examination; FDG, fluorodeoxyglucose; pg, picogram; ml, milliliter; mm, millimeter. Data are expressed in mean±standard deviation (SD) or frequency (%). aCSF Aβ42 data were available for 191 subjects and 93 PiB-PET-positive. bSUVR data were available for 191 subjects and 108 PiB-PET-positive. c1CSF t-tau data were available for 194 subjects and 95 PiB-PET-positive. c2CSF p-tau data were available for 268 subjects and 122 PiB-PET-positive. dCDR-SB data were available for 232 subjects and 108 PiB-PET-positive. e1MMSE baseline data were available for 231 subjects and 107 PiB-PET-positive. e2MMSE last follow-up were available for 232 subjects and 108 PET-positive. f1FDG-PET data at baseline were available for 196 subjects and 102 PET-positive. f2FDG-PET at last visit were available for 172 subjects and 92 PET-positive. g1MRI baseline data were available for 215 subjects and 108 PET-positive. g2MRI last visit were available for 200 subjects and 98 PET-positive.
Primary endpoint
Amyloid PiB-PET-positive cohort
A total of 43 (39.8%) subjects met criteria for CDR progression. In adjusted analyses, the risk of progression was reduced to a greater extent by higher CSF Aβ42 (RR, 0.36; 95% CI, 0.19–0.67; p = 0.002) than lower SUVR (RR, 0.81; 95% CI, 0.68–0.96; p = 0.018) but not than lower t-tau or lower p-tau (Fig. 1, top). These results were unchanged after excluding 5% of the SUVR threshold. In addition, higher CSF Aβ42 levels were associated with a reduced hazard of time to first CDR progression (HR, 0.37; 95% CI, 0.18–0.77; p = 0.008) to a greater extent than lower SUVR (HR, 0.79; 95% CI, 0.62–1.02; p = 0.075) and lower p-tau (HR, 0.63; 95% CI, 0.40–0.98; p = 0.041). These results were confirmed in the stratified Cox model (Supplementary Table 2). CSF Aβ42 levels predicted progression to a greater extent than SUVR, CSF t-tau, or CSF p-tau levels in all analyses excluding p-tau or t-tau from the models to address multicollinearity, after additionally adjusting for baseline CDR and age, or after missing imputations (Supplementary Tables 3 and 4).
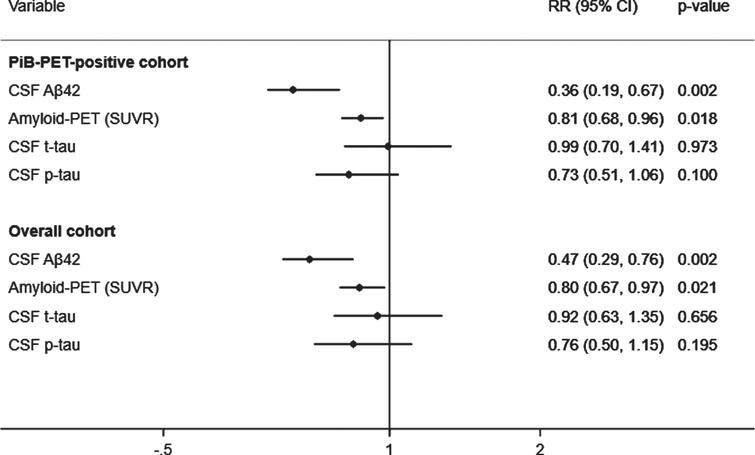
Adjusted prediction of CDR progression with baseline CSF Aβ42, p-tau, t-tau, and amyloid (PiB)-PET SUVR levels. CSF, cerebrospinal fluid; PiB-PET, Pittsburgh compound B positron emission tomography; SUVR, standardized uptake value ratio; CDR, Clinical Dementia Rating; Aβ42, 42-amino acid amyloid-beta peptide; p-tau, phospho-Tau; t-tau, total-Tau; HR, hazard ratio; CI, confidence interval. RR reflects effect size for the association with a one standard deviation higher in CSF Aβ42levels and lower in SUVR, CSF t-tau and p-tau levels. Analyses were adjusted for age at onset, sex, education, APOE4, and duration of follow up. Overall cohort includes PiB-PET-positive and negative samples.
Overall cohort
A total of 67 (28.9%) subjects met criteria for CDR progression in the overall cohort. Higher CSF Aβ42 levels predicted a reduced risk of progression (RR, 0.47; 95% CI, 0.29–0.76; p = 0.002) to a greater extent than lower SUVR (RR, 0.80; 95% CI, 0.67–0.97; p = 0.021), but not than lower t-tau or p-tau (Fig. 1, bottom). The results were unchanged after additionally adjusting for baseline CDR and age in all analyses with or without p-tau or t-tau or after missing imputations (Supplementary Tables 3 and 4). Moreover, higher CSF Aβ42 levels were associated a reduced hazard of time to first CDR progression (HR, 0.51; 95% CI, 0.31–0.86; p = 0.011) than lower SUVR (HR, 0.79; 95% CI, 0.62–1.01; p = 0.062) and lower p-tau (HR, 0.62; 95% CI, 0.40–0.97; p = 0.037). These results were confirmed in the stratified Cox model (Supplementary Table 2).
CSF Aβ42 and CDR progression
CSF Aβ42 levels were higher among CDR non-progressors than CDR progressors (Fig. 2). CSF Aβ42 <270 pg/ml predicted progression (area under the curve, 80.5%; sensitivity, 72.2%; specificity, 74.5%) regardless of increasing SUVR levels. The progression-free survival was longer with CSF Aβ42 ≥270 pg/ml compared to CSF Aβ42 <270 pg/ml over the follow-up period in both PiB-PET-positive (p = 0.002) and overall cohorts (p < 0.001) (Supplementary Figure 2). In adjusted analysis, higher CSF Aβ42 levels reduced the risk of progression even at very high PiB-PET SUVR levels. CSF Aβ42 levels predicting lower risk of progression increased with higher SUVR levels (Fig. 3).
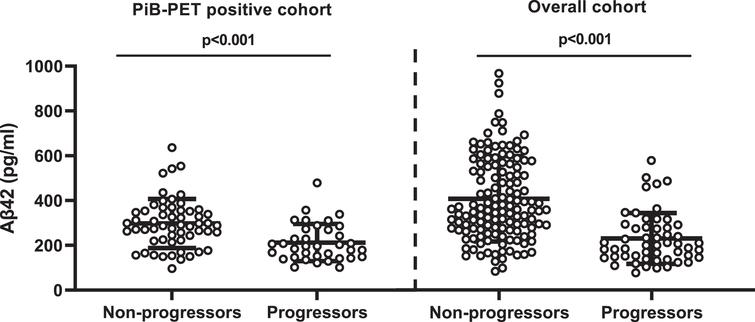
Comparison of CSF Aβ42 levels between non-progressors and progressors. PiB-PET-positive cohort: non-CDR progressors (297.73±13.66) versus CDR progressors (218.73±17.22); overall cohort: non-CDR progressors (380.83±14.5) versus CDR progressors (313.35±26.46). Error bar represents the standard error of mean. CSF, cerebrospinal fluid; PiB-PET, Pittsburgh compound B positron emission tomography; CDR, Clinical Dementia Rating; Aβ42, 42-amino acid amyloid-beta peptide. Overall cohort includes PiB-PET-positive and negative samples.
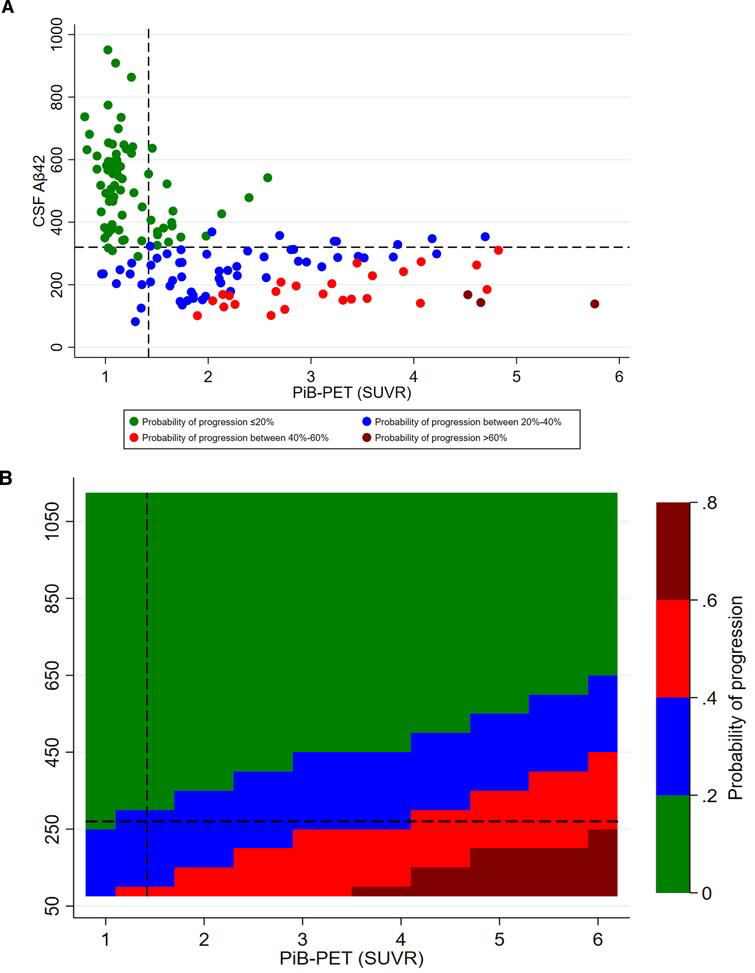
Adjusted probability of CDR progression. A) scatter plot of CSF Aβ42 and PiB-PET SUVR levels; B) contour plot of CSF Aβ42 and PiB-PET SUVR levels. CSF, cerebrospinal fluid; Aβ42, 42-amino acid amyloid-beta peptide; PiB-PET, Pittsburgh compound B positron emission tomography; CDR, Clinical Dementia Rating; p-tau, phospho-Tau; t-tau, total-Tau. All models were adjusted for age at onset, sex, education, APOE4, p-tau, and t-tau levels. Overall cohort includes PiB-PET-positive and negative samples.
Secondary endpoints
Amyloid PiB-PET-positive cohort
Higher baseline CSF Aβ42 predicted a reduced risk of progression to CDR ≥0.5 (RR, 0.55; 95% CI, 0.36–0.83; p = 0.004) and to CDR ≥1 (RR, 0.27; 95% CI, 0.13–0.59; p = 0.001), and was associated with CDR-SB ≥4.5 (RR, 0.36; 95% CI, 0.36–0.18–0.70; p = 0.003) and MMSE ≤24 (RR, 0.30; 95% CI, 0.17–0.54; p < 0.001) better than lower SUVR, lower t-tau, or lower p-tau levels (Table 2). Higher CSF Aβ42 levels were also associated with larger hippocampi volume and higher FDG-PET uptake at baseline to a greater extent than lower SUVR, lower p-tau or lower t-tau levels (data not shown). These results were unchanged in age-adjusted models with other covariates and after removing t-tau and p-tau from the models (Supplementary Tables 5 and 6). Higher baseline CSF Aβ42 levels were associated with larger hippocampal volume (RC, 319.91; 95% CI, 73.97–565.86; p = 0.012) and higher FDG-PET uptake (RC, 0.14; 95% CI, 0.03–0.24; p = 0.011) to a greater extent than lower PiB-PET SUVR levels (Supplementary Table 7). The results were unchanged after excluding 5% of the SUVR threshold and in age-adjusted models with other covariates (Supplementary Table 7). The association between CSF Aβ42 levels and all secondary endpoints remained significant after excluding t-tau or p-tau (Supplementary Table 8).
| PiB-PET-positive cohort | PiB-PET-positive cohort (SUVR ≥1.49) | Overall cohort | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RR* | 95% CI | p | RR* | 95% CI | p | RR* | 95% CI | p | ||||
| Progression to CDR ≥0.5 | ||||||||||||
| CSF Aβ42 | 0.55 | 0.36 | 0.83 | 0.004 | 0.59 | 0.38 | 0.91 | 0.017 | 0.52 | 0.35 | 0.77 | 0.001 |
| SUVR (PiB-PET) | 0.75 | 0.63 | 0.90 | 0.002 | 0.78 | 0.65 | 0.93 | 0.006 | 0.70 | 0.59 | 0.84 | <0.001 |
| CSF t-tau | 0.97 | 0.78 | 1.19 | 0.764 | 0.96 | 0.78 | 1.19 | 0.693 | 0.98 | 0.78 | 1.23 | 0.876 |
| CSF p-tau | 0.67 | 0.52 | 0.86 | 0.002 | 0.69 | 0.53 | 0.89 | 0.005 | 0.60 | 0.46 | 0.79 | <0.001 |
| Progression to CDR ≥1 | ||||||||||||
| CSF Aβ42 | 0.27 | 0.13 | 0.59 | 0.001 | 0.29 | 0.13 | 0.62 | 0.002 | 0.26 | 0.13 | 0.51 | <0.001 |
| SUVR (PiB-PET) | 0.72 | 0.55 | 0.94 | 0.016 | 0.74 | 0.56 | 0.97 | 0.028 | 0.66 | 0.51 | 0.87 | 0.003 |
| CSF t-tau | 0.95 | 0.74 | 1.23 | 0.724 | 0.94 | 0.74 | 1.22 | 0.681 | 0.96 | 0.74 | 1.25 | 0.751 |
| CSF p-tau | 0.57 | 0.36 | 0.89 | 0.015 | 0.59 | 0.37 | 0.92 | 0.020 | 0.50 | 0.32 | 0.78 | 0.002 |
| CDR-SB ≥4.5 at last visit# | ||||||||||||
| CSF Aβ42 | 0.36 | 0.18 | 0.70 | 0.003 | 0.38 | 0.19 | 0.74 | 0.005 | 0.32 | 0.18 | 0.58 | <0.001 |
| SUVR (PiB-PET) | 0.78 | 0.63 | 0.96 | 0.018 | 0.79 | 0.64 | 0.98 | 0.030 | 0.73 | 0.59 | 0.90 | 0.004 |
| CSF t-tau | 1.09 | 0.85 | 1.39 | 0.492 | 1.09 | 0.85 | 1.37 | 0.520 | 1.12 | 0.88 | 1.43 | 0.366 |
| CSF p-tau | 0.47 | 0.29 | 0.75 | 0.002 | 0.48 | 0.30 | 0.76 | 0.002 | 0.40 | 0.25 | 0.64 | <0.001 |
| MMSE ≤24 at last visit# | ||||||||||||
| CSF Aβ42 | 0.30 | 0.17 | 0.54 | <0.001 | 0.32 | 0.18 | 0.59 | <0.001 | 0.26 | 0.15 | 0.45 | <0.001 |
| SUVR (PiB-PET) | 0.81 | 0.64 | 1.03 | 0.094 | 0.83 | 0.66 | 1.06 | 0.135 | 0.75 | 0.59 | 0.96 | 0.022 |
| CSF t-tau | 0.98 | 0.69 | 1.39 | 0.931 | 0.97 | 0.70 | 1.35 | 0.872 | 1.02 | 0.72 | 1.45 | 0.921 |
| CSF p-tau | 0.61 | 0.40 | 0.94 | 0.026 | 0.64 | 0.42 | 0.97 | 0.036 | 0.52 | 0.34 | 0.81 | 0.003 |
Adjusted associations of baseline CSF and SUVR with secondary cognitive outcomes
*Relative risk (RR) is with a one standard deviation higher in CSF Aβ42 levels and lower in CSF t-tau, CSF p-tau, and SUVR levels. CI, confidence interval; CDR, clinical dementia rating; CSF, cerebrospinal fluid; Aβ42, 42-amino acid amyloid-beta peptide; t-tau, total tau; p-tau, phospho-Tau; CDR-SB, CDR sum of boxes; PiB-PET, Pittsburgh compound B positron emission tomography; SUVR, standardized uptake value ratio; MMSE, Mini-Mental State Examination; FDG, fluorodeoxyglucose. All CSF and SUVR values are standardized. Analysis adjusted for mean mutation age of symptom onset, sex, education, APOE4 status, and duration of follow up. #Analyses were also adjusted for baseline CDR-SB or MMSE; Overall cohort includes PiB-PET-positive and negative samples.
Overall cohort
Higher baseline CSF Aβ42 predicted a reduced risk of progression to CDR ≥0.5 (RR, 0.52; 95% CI, 0.35, 0.77; p = 0.001) and to CDR ≥1 (RR, 0.26; 95% CI, 0.13, 0.51; p < 0.001), and was associated with CDR-SB ≥4.5 (RR, 0.32; 95% CI, 0.18, 0.58; p < 0.001) and MMSE ≤24 (RR, 0.26; 95% CI, 0.15, 0.45; p < 0.001) better than lower SUVR, lower t-tau, or lower p-tau levels (Table 2). Similar results were observed in age-adjusted models with other covariates (Supplementary Table 5) and models with and without p-tau or t-tau (Supplementary Table 6). Larger hippocampal volume was inversely associated with SUVR levels (RC, –204.70; 95% CI, –341.07, –68.33; p = 0.004) and p-tau levels (RC, –219.06; 95% CI, –405.74, –32.37; p = 0.022) but not with CSF Aβ42 levels (Supplementary Table 7). Higher SUVR was associated with lower FDG-PET uptake (RC, –0.07; 95% CI, –0.13, –0.01; p = 0.022) but not with CSF Aβ42 (Supplementary Tables 7 and 8).
DISCUSSION
This longitudinal analysis of a genetic cohort showed that in amyloid PiB-PET-positive individuals with autosomal dominant AD-causing mutations higher levels of CSF Aβ42 were associated with reduced risk of progression to cognitive impairment, as well as larger hippocampal volume and higher brain metabolism in the precuneus. Conversely, cognitive deterioration was strongly predicted by lower levels of soluble Aβ42 but not by increases in SUVR, t-tau, or p-tau. These data are in agreement with those of a cross-sectional analysis of a sporadic AD cohort (in participants of the Alzheimer’s Disease Neuroimaging Inventory cohort study), in which higher CSF Aβ42 levels were associated with normal cognition regardless of (and despite increasing) SUVR levels among PiB-PET-positive individuals [11], making a reduction in CSF Aβ42 levels a better marker of progression than higher levels of CSF t-tau, p-tau, or amyloid PiB-PET SUVR.
A threshold of compensation can tentatively be drawn from these data, with further studies needed to validating it. We found that levels of CSF Aβ42 above 270 pg/ml predicted a reduced risk of CDR progression across any levels of PiB-PET SUVR. The better prediction of disease conversion by the reduction of CSF Aβ42 levels than by the increase in brain amyloidosis is in line with its observed decline as early as 25 years before the onset of cognitive impairment in genetic AD [4]. There is also support for a greater pathophysiologic importance of the loss of soluble Aβ42 over the accrual of insoluble PiB-PET amyloid from knockout animal models of the Aβ42 precursor, AβPP, which yields neurodegeneration without brain amyloidosis [20], and in AD patients treated with BACE-1 inhibitors, which reduced CSF Aβ42 levels and worsened cognitive symptoms regardless of changes in brain amyloid [21]. In vitro studies have shown that PSEN mutations reduce the levels of Aβ42 and Aβ40 [22], even if some mutations increase the Aβ42/40 ratio [23, 24]. Clinically, however, mutation carriers, including Down syndrome patients with APP duplications, have lower CSF levels of Aβ42 and Aβ42/40 ratio at the symptomatic stage compared to cognitively unimpaired individuals [3]. Supporting the importance of the soluble protein fraction, individuals with dementia associated with the rare APP E693del (Osaka) mutation exhibit low CSF Aβ42 despite no PiB-PET-amyloid positivity [25]. Importantly, the inverse correlation between the progressive loss of soluble Aβ42 along with the increase in insoluble Aβ is imperfect. In fact, we previously showed that soluble Aβ42 levels can still be high even in individuals with the highest amyloid-SUVR PiB-PET tertile, a relationship associated with normal cognition [11].
Amyloids represent the end product of an irreversible phase transition process of proteins, changing from a soluble to an insoluble state via a physicochemical process termed nucleation [26]. Nucleation is the rate-limiting step of the first stable amyloid cluster (nucleus), after which the phase transition proceeds spontaneously until all the available soluble substrate is consumed. While nucleation is not favorable under normal conditions, it becomes favorable at higher concentrations, such as those attained via gene duplication, or when the protein is rendered unstable due to structural mutations [27]. The net outcome is decreased availability of soluble Aβ42. This explains why their depletion is universal across all reported familial AD mutations, including APP duplications [3, 28]. Whereas an increase in amyloid plaque burden in the brain exhibits an inconsistent relationship with cognitive impairment, the depletion of soluble Aβ42 is universally associated with dementia in all familial and sporadic forms of AD, further suggesting it is pathophysiologically more relevant [1, 3, 4, 29]. This is supported by an extensive body of literature demonstrating the role of physiological (picomolar) concentrations of Aβ42 in memory and synaptic plasticity via alpha-7 nicotinic acetylcholine receptor (α7-nAChR) signaling (Supplementary Table 9). Very recently, synthetic Aβ42 monomers have been shown to improve impaired memory in conditional double knockout mice without plaque deposition and in the APP/PS1/Tau triple transgenic mice with Aβ42 deposition, effects that were mediated by the α7-nAChR [30]. These results not only indicate the importance of the loss of Aβ42 function as a pathogenic mechanism both in the presence and absence of plaques, but also suggest that Aβ42replacement has therapeutic potential.
Major strengths of this analysis are its longitudinal design and the relatively large number of subjects with disease-causing mutations. The findings are supported by the use of several complementary analytic methods on cognitive assessments and neuroimaging data and confirmed by sensitivity analyses. Major limitations include the inability to adjust for the estimated year to symptom onset (highly correlated with CSF and SUVR levels and did not serve as a confounder), and the lack of data on CSF Aβ40 (and therefore of Aβ42/Aβ40 ratio; see below) and on oligomeric species. On the last point, subjects with normal cognition despite high PiB-PET plaque burden had high, not low CSF Aβ42 [11], opposite to the direction predicted by the hypothesis of oligomeric toxicity. Oligomers are very transient and the majority of them dissociate back to monomers rather than progress to aggregates [31]. Importantly, the reduction of soluble Aβ42 levels during the disease course reduces the substrate for the oligomers, further questioning any sustainable toxicity from them.
A note related to the issue of ratio versus absolute values is worth making. Early studies argued that absolute levels of Aβ42 decrease but the Aβ42/Aβ40 ratio increases due to the lower decrease of Aβ40 relative the more aggregation-prone Aβ42 [32, 33]. However, recent studies have found that the Aβ42/Aβ40 ratio decreases in AD [3, 34]. Pathophysiologically, amyloid aggregation is dependent on supersaturation [35–37], which is based on the absolute concentration of the peptide. Decreasing peptide concentration will decrease, not increase, the saturation and the related propensity to form any type of aggregates irrespective of its relative levels compared to other peptides [38]. Nearly 90% of AD-related mutations in the PSEN1 gene lead to reduced production of both Aβ42 and Aβ40 [22], a classical genetic loss-of-function mechanism. Further, there is no correlation between the Aβ42/Aβ40 ratio and the age of onset in mutation carriers [22].
In conclusion, higher soluble Aβ42 levels are associated with reduced risk of CDR progression, normal cognition, normal hippocampal volume, and normal precuneus metabolism to a greater extent than lower brain amyloid, lower p-tau, and lower t-tau levels in amyloid PiB-PET-positive individuals with autosomal dominant AD-causing genetic mutations. Brain toxicity in AD may be predominantly mediated by a reduction of the soluble protein pool, its functional fraction, rather than its accrual into amyloids.