
Earlier Alzheimer’s disease onset is associated with tau pathology in brain hub regions and facilitated tau spreading
By Lukas Frontzkowski, Michael Ewers, Matthias Brendel, Davina Biel, Rik Ossenkoppele, Paul Hager, Anna Steward, Anna Dewenter, Sebastian Römer, Anna Rubinski, Katharina Buerger, Daniel Janowitz, Alexa Pichet Binette, Ruben Smith, Olof Strandberg, Niklas Mattsson Carlgren, Martin Dichgans, Oskar Hansson, and Nicolai Franzmeier
Excerpt from the article published in Nature Communications 13, 4899, 20 August 2022, DOI: https://doi.org/10.1038/s41467-022-32592-7
Editor’s Highlights
- In sporadic Alzheimer’s disease (AD), younger age of symptom onset has been previously associated with accelerated tau accumulation and neurodegeneration, faster cognitive decline and higher mortality, together suggesting that younger symptom onset is potentially driven by a more aggressive form of AD.
- Younger symptomatic manifestation of AD show stronger tau pathology in globally connected hubs in the fronto-parietal association cortex compared to inferior temporal non-hubs, which may drive faster tau spreading and accelerated cognitive decline.
- This supports previous findings of tau-pathology being a key driver of symptomatic disease manifestation in AD, while adding important new evidence that stronger tau deposition in globally connected hubs is associated with earlier AD symptom manifestation.
- Importantly, these associations were not detected for amyloid-positive, yet asymptomatic individuals, in whom tau pathology was mostly absent.
Abstract
In Alzheimer’s disease (AD), younger symptom onset is associated with accelerated disease progression and tau spreading, yet the mechanisms underlying faster disease manifestation are unknown. To address this, we combined resting-state fMRI and longitudinal tau-PET in two independent samples of controls and biomarker-confirmed AD patients (ADNI/BioFINDER, n = 240/57). Consistent across both samples, we found that younger symptomatic AD patients showed stronger tau-PET in globally connected fronto-parietal hubs, i.e., regions that are critical for maintaining cognition in AD. Stronger tau-PET in hubs predicted faster subsequent tau accumulation, suggesting that tau in globally connected regions facilitates connectivity-mediated tau spreading. Further, stronger tau-PET in hubs mediated the association between younger age and faster tau accumulation in symptomatic AD patients, which predicted faster cognitive decline. These independently validated findings suggest that younger AD symptom onset is associated with stronger tau pathology in brain hubs, and accelerated tau spreading throughout connected brain regions and cognitive decline.
Introduction
Sporadic Alzheimer’s disease (AD) is highly heterogeneous, with variable clinical expression, age of symptom onset as well as cognitive and neuropathological trajectories1,2. In sporadic AD, younger age of symptom onset has been previously associated with accelerated tau accumulation3,4 and neurodegeneration5, faster cognitive decline6,7,8 and higher mortality9, together suggesting that younger symptom onset is potentially driven by a more aggressive form of AD9,10. Yet, it is unclear why sporadic AD patients with younger symptom onset show faster pathological3,4,5 and clinical progression6,7,8 than patients with later symptom onset. From a histopathological point of view, non-mendelian early- and late-onset sporadic AD share the same molecular pathologies including amyloid-beta (Aβ) plaques and neurofibrillary tangles composed of hyperphosphorylated 3/4R tau pathology, suggesting that an earlier symptom onset is unlikely to be specifically driven by distinct molecular characteristics of AD pathology11. Rather, previous post-mortem and tau-PET imaging studies reported that an earlier AD symptom onset is associated with a different spatial distribution pattern of tau pathology10,12, i.e., the key driver of neurodegeneration13 and cognitive decline in AD14,15. Specifically, post-mortem assessments in AD patients found that a more pronounced neocortical and hippocampal-sparing pattern of neurofibrillary tau tangle pathology is associated with younger age at symptom onset and faster ante-mortem cognitive decline, whereas a spatially more restricted limbic-predominant pattern of neurofibrillary tau tangles was associated with older age at symptom onset and slower ante-mortem cognitive decline10. Similarly, tau-PET studies in AD patients found that younger age in general12,16 and early-onset AD (i.e., before the age of 65) in particular are associated with stronger tau-PET uptake in fronto-parietal association cortex regions12,17,18,19 with relative sparing of medial temporal lobe regions18. Together, these findings suggest that an earlier symptom onset in AD is associated with a pattern shift of tau pathology deposition from allocortical medial temporal lobe regions toward neocortical fronto-parietal association cortices.
As shown by functional MRI studies, fronto-parietal brain regions harbor key hubs that are globally connected to the rest of the brain20,21 and central for cognitive function22,23 as they facilitate information integration across different brain networks during cognitive demands22. In a series of resting-state fMRI studies, we and others could previously show that the functional integrity of fronto-parietal control network hubs is critically important for maintaining cognitive function in aging24, AD25,26,27 and other neurodegenerative diseases28. Since tau pathology has been shown to impair neuronal function29 and to drive neurodegeneration13, a stronger tau pathology load in fronto-parietal hub regions that are highly relevant for cognition may thus drive earlier symptom manifestation in AD. In addition, tau pathology deposition in globally connected hub regions may further accelerate the progression and spreading of tau pathology itself, which is in turn a strong driver of cognitive decline30. Specifically, preclinical studies have consistently shown that tau spreads trans-synaptically across interconnected neurons31,32. Similarly, we and others reported previously in combined tau-PET and MRI studies that tau spreads preferentially across functionally and anatomically connected brain regions, where the connectivity pattern of tau harboring epicenter regions determines the subsequent spreading pattern of tau pathology2,33,34,35,36,37,38. Thus, the occurrence of tau pathology in globally connected hubs early in the course of AD may ensue faster and more widespread tau spreading thereby driving earlier disease manifestation and faster clinical progression14,15,30.
To address these open questions, the major aims of the current combined tau-PET and resting-state fMRI study were to assess whether: (1) younger age is associated with stronger tau pathology in globally connected hub regions compared to non-hubs in patients with symptomatic AD; (2) whether tau pathology in hub regions is associated with accelerated subsequent tau spreading and; (3) whether the association between younger age and faster tau accumulation rates in symptomatic AD3 is mediated by stronger tau pathology in globally connected hub regions. To this end, we employed two independent samples covering the entire AD spectrum, including 240 participants of the Alzheimer’s disease neuroimaging initiative (ADNI) and 57 subjects of the BioFINDER study for replication, all with available baseline amyloid-PET and longitudinal Flortaucipir tau-PET. To map the topology of globally connected hubs across the brain, we used high-resolution resting-state fMRI data from 1000 healthy participants of the human connectome project (HCP). Based on these resting-state fMRI data, we estimated an atlas-based39 whole-brain map of the graph-theoretical metric weighted degree (i.e., also referred to as global connectivity)25 in healthy individuals that are unaffected by AD pathology. By mapping individual tau-PET patterns in AD patients to the topology of globally connected brain hubs, we determined the degree to which individual tau-PET patterns are expressed in globally connected hub regions in the fronto-parietal association cortex vs. weakly connected non-hub regions e.g., in temporo-limbic and visual cortex region. Based on these data, we show that (1) individual tau-PET deposition patterns are indeed stronger in globally connected hub regions in younger patients with symptomatic AD and associated with earlier symptom onset; (2) that a more hub-like pattern of tau pathology deposition at baseline is associated with accelerated subsequent annual tau accumulation and; (3) that the association between younger age and faster tau accumulation rates in symptomatic AD is mediated by a more hub-like tau-PET pattern thereby driving faster cognitive decline.
Results
To address and validate the aims of the current study, we included two fully independent samples (ADNI and BioFINDER) with baseline amyloid-PET and longitudinal 18F-Flortaucipir tau-PET. We further included longitudinal cognitive data for a memory composite as well as age of symptom onset estimates in symptomatic AD patients which were available for a subset in ADNI. Both samples included cognitively normal (CN) control subject without evidence of AD pathology (ADNI/BioFINDER, n = 93/16), CN amyloid-positive individuals (i.e., preclinical AD, ADNI/BioFINDER, n = 60/16) and patients with symptomatic AD (i.e., mild cognitively impaired (MCI) and AD dementia; ADNI/BioFINDER, n = 89/25). Subject demographics for both samples are displayed in Table 1. Resting-state fMRI data from 1000 HCP participants was used to estimate a healthy connectivity template for the Schaefer 200 ROI atlas that covers the neocortex (Fig. 1A)39. Across the 1000 HCP participants, we created a group-average functional connectivity matrix, which was density thresholded at 30% to eliminate potentially weak and noisy connections and transformed to connectivity-based distance (i.e., higher functional connectivity = shorter connectivity-based distance, Fig. 1B). For each ROI, we then determined the mean connectivity-based distance to the remaining ROIs as a measure of hub-ness (i.e., shorter distance to the rest of the brain = higher hub-ness, Fig. 1C), which is equivalent to the graph theoretical measure weighted degree. In line with previous studies20,40, hub regions were primarily found in the fronto-parietal association cortex. To determine a hub map that scales brain regions between hubs and non-hubs, the group-average global connectivity map (Fig. 1C) was rescaled between 1 (i.e., hub) and −1 (i.e., non-hub) (Fig. 1D), in order to assess in a later step whether individual tau-PET patterns matched a hub or non-hub pattern. Tau-PET SUVR data (i.e., intensity normalized to the inferior cerebellar gray) was parcellated using the same 200 ROI parcellation (Fig. 1A), and pre-established two-component Gaussian mixture modeling (Fig. 1E) was applied to transform tau-PET SUVRs to tau-PET positivities in order to separate off-target from on-target tau-PET binding33. In an additional exploratory step, we also included tau-PET SUVRs from pre-established hippocampal ROIs41, which exclude the choroid plexus and therefore minimize the influence of Flortaucipir off-target binding. As for the remaining cortex, hippocampal tau-PET SUVRs underwent two-component Gaussian mixture modeling to further reduce the effect of off-target binding. Surface renderings of baseline tau-PET positivity across diagnostic groups are shown in Fig. 1F for ADNI and for BioFINDER, showing no evidence for tau-PET positivity in amyloid-negative controls, minimally elevated temporal-lobe tau levels in preclinical AD, vs. gradually increasing tau-PET positivity across symptomatic AD patients with MCI and AD dementia.
| ADNI (n = 240) | CN Aβ− | CN Aβ+ | MCI Aβ+ | AD dementia | p value |
| (n = 93) | (n = 60) | (n = 56) | (n = 31) | ||
| Age (M/SD) | 72.54 (6.84) | 75.23 (6.65) | 75.14 (7.63) | 76.87 (7.74) | 0.011 |
| Sex (m/f) | 39/54 | 26/34 | 29/27 | 20/11 | 0.134 |
| Education (M/SD) | 16.51 (2.48) | 16.6 (2.24) | 16.25 (2.56) | 15.9 (2.68) | 0.568 |
| MMSE (M/SD) | 29.21 (0.91) | 28.78 (1.78) | 27.33 (1.92) | 21.75 (5.44) | <0.001 |
| ADNI-MEM (M/SD) | 1.11 (0.59) | 0.87 (0.53) | 0.2 (0.63) | −0.77 (0.66) | <0.001 |
| Centiloid (M/SD) | −3.62 (12.88) | 69.62 (32.63) | 76.72 (29.98) | 89.28 (36.33) | <0.001 |
| ApoE4 (pos/neg/NA) | 29/50/14 | 33/21/6 | 32/10/14 | 12/6/13 | <0.001 |
| Global tau-PET SUVR | 1.08 (0.08) | 1.10 (0.09) | 1.21 (0.22) | 1.38 (0.41) | <0.001 |
| Temporal meta ROI tau-PET SUVR | 1.12 (0.1) | 1.15 (0.1) | 1.29 (0.23) | 1.43 (0.36) | <0.001 |
| Tau hub gradient (M/SD) | −0.12 (0.16) | −0.17 (0.17) | −0.16 (0.19) | −0.08 (0.24) | 0.090 |
| Mean tau-PET follow-up time in years (M/SD) | 2.14 (1.12) | 1.86 (0.71) | 1.70 (0.83) | 1.62 (0.61) | <0.001 |
| BioFINDER (n = 57) | CN Aβ− | CN Aβ+ | MCI Aβ+ | AD dementia | p value |
| (n = 16) | (n = 16) | (n = 7) | (n = 18) | ||
| Age | 73.88 (5.32) | 75.44 (6.09) | 72.71 (6.63) | 69.83 (10.48) | 0.192 |
| Sex (m/f) | 10/6 | 6/10 | 2/5 | 11/7 | 0.245 |
| Education (M/SD) | 12.59 (4.06) | 10.56 (3.22) | 11.14 (2.67) | 13.44 (3.26) | 0.097 |
| MMSE (M/SD) | 29 (1.1) | 29.31 (1.08) | 25.57 (2.94) | 22.06 (5.17)b | <0.001 |
| Global Flutemetamol SUVR | 0.52 (0.03) | 0.77 (0.12) | 0.84 (0.14) | 0.97 (0.15) | <0.001 |
| ApoE4 (pos/neg/NA) | 0/16/0 | 10/6/0 | 4/3/0 | 10/7/1 | <0.001 |
| Global tau-PET SUVR | 1.04 (0.05) | 1.05 (0.05) | 1.37 (0.37) | 1.49 (0.36) | <0.001 |
| Temporal meta ROI tau-PET SUVR | 1.08 (0.06) | 1.09 (0.05) | 1.52 (0.39) | 1.56 (0.31) | <0.001 |
| Tau hub gradient (M/SD) | −0.16 (0.22) | −0.09 (0.23) | −0.15 (0.23) | −0.06 (0.20) | 0.566 |
| Mean tau-PET follow-up time in years (M/SD) | 2.03 (0.47) | 1.91 (0.32) | 1.82 (0.12) | 1.97 (0.34) | 0.484 |
Subject demographics
p values were derived from ANOVAs for continuous measure sand from Chi-squared tests for categorical measures.
M mean, SD standard deviation, m male, f female, MMSE mini-mental state exam, SUVR standardized uptake value ratio, NA not available.
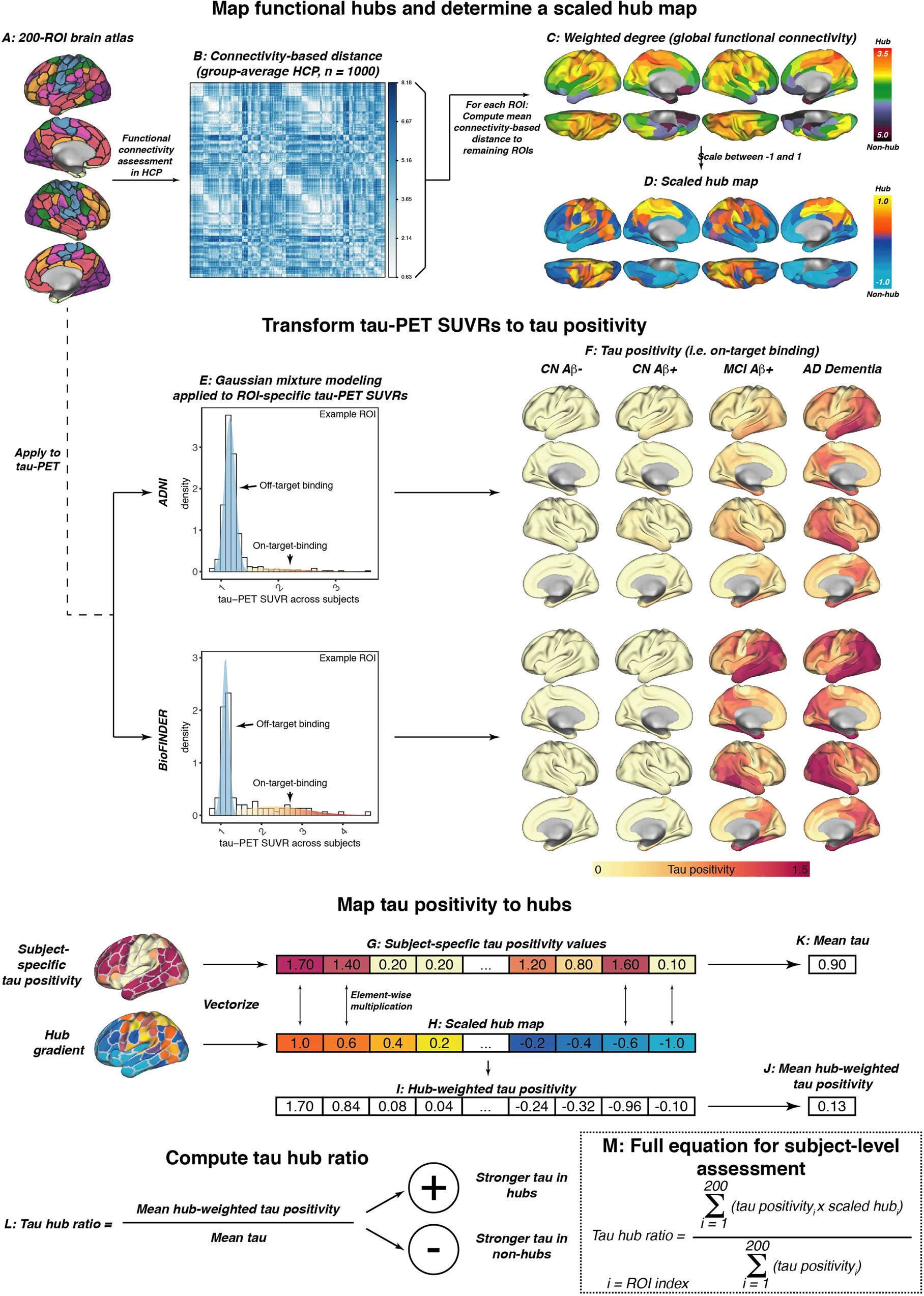
fMRI and tau-PET processing.
A Surface rendering of the 200 region of interest (ROI)-brain atlas, based on which we estimated (B) inter-regional functional connectivity-based distance based on 1000 participants of the human connectome project. Specifically, subject-specific connectivity matrices (i.e., Fisher-z transformed correlations of ROI-specific preprocessed fMRI timeseries) were averaged across subjects, thresholded at a density of 30% (i.e., only the strongest 30% of connections were retained) and transformed to connectivity-based distance (i.e., shorter distance = higher connectivity). C For each ROI, we determined the average connectivity-based distance to the remaining 199 ROIs as a measure of weighted degree (i.e., global connectivity), which was subsequently rescaled between −1 and 1 to determine a (D) continuous mapping of connectivity from hubs to non-hubs. The same brain atlas shown in A was applied to tau-PET standardized uptake value ratios (SUVRs), and E two-component Gaussian mixture modeling was applied to transform tau-PET SUVRs to tau positivities, i.e., tau-PET SUVRs that have been cleaned from the off-target binding curve. F Surface renderings of group-average tau positivities are shown for each diagnostic group of the ADNI and BioFINDER sample. G Subject-specific tau positivity values were subsequently multiplied with the (H) scaled hub map to determine (I) hub-weighted tau positivity values, giving tau in hub regions a positive weight and tau in non-hub regions a negative weight. J Hub-weighted tau positivities and K tau positivities were subsequently averaged in order to (L) compute the tau hub ratio, i.e., a single numeric index indicating whether an individual tau PET pattern was more pronounced in hub regions (i.e., more positive) or non-hub regions (i.e., more negative), while adjusting for global tau levels. The mathematical equation for determining the tau hub ratio for subject-level data is shown in M. Source data are provided as a Source Data file.
Younger age is associated with a more hub-like tau-PET pattern in symptomatic AD
To assess the degree to which individual tau-PET deposition represented a hub or non-hub pattern, we mapped patient-specific tau-PET positivity to the scaled hub map shown in Fig. 1D. Specifically, we multiplied ROI-specific subject-specific tau-positivity with the scaled hub map shown in Fig. 1D, in order to determine hub-weighted tau positivity values, which were subsequently averaged and divided by the overall tau-PET positivity averaged across all 200 ROIs, to adjust for global tau deposition. We specifically adjusted for global tau levels, so that the tau hub ratio specifically reflected the pattern of tau deposition and not the overall tau severity. The mapping of subject-specific tau-PET patterns to the scaled hub map is illustrated in Fig. 1G–M, yielding a numeric index (i.e., henceforth referred to as tau hub ratio) that indicates whether a subject-specific tau-PET pattern is more representative of hub regions (i.e., more positive) or non-hub regions (i.e., more negative), while adjusting for global tau-PET positivity. Using an ANOVA, no difference in the tau hub ratio was found between diagnostic groups (Table 1, ADNI, p = 0.090, BioFINDER, p = 0.566), highlighting that the tau hub ratio does capture spatial patterns of tau deposition but not increasing diagnostic severity. We then tested whether younger age was associated with a higher tau hub ratio in patients with symptomatic AD (i.e., MCI and dementia), using linear regression controlling for sex, education and diagnosis. Supporting this, we found younger age to be associated with a more positive tau hub ratio in symptomatic AD patients of both samples (ADNI: β = −0.238, p = 0.024, Fig. 2A, right panel; BioFINDER: β = −0.482, p = 0.018, Fig. 2B, right panel). Exact tests using 1000 beta values from null-model tau hub ratios that were generated using shuffled connectomes as a reference confirmed these results (ADNI: p = 0.016; BioFINDER: p = 0.019). Further, in a subset of 38 symptomatic ADNI participants with available resting-state fMRI data, this association was consistent when using subject-level connectivity data and tau-PET to determine the tau hub ratio (b = −0.307, p = 0.037). Results also remained significant when repeating the analyses using robust regression or when additionally controlling for global Aβ levels or ApoE4 status (only available in a subset, see Table 1 and see Supplementary Table 1). In contrast, no association between age and the tau hub ratio was detected for asymptomatic patients with preclinical AD (ADNI: β = 0.087, p = 0.542, Fig. 2A, left panel; BioFINDER: β = −0.307, p = 0.901, Fig. 2B, left panel), in which abnormal tau-PET signal was minimal. When repeating the above-described analysis with the actual age of cognitive symptom onset based on informant assessments (i.e., available in 77/89 symptomatic ADNI participants), we found congruent results, showing that younger age of symptom onset was associated with a higher tau hub ratio (β = −0.256, p = 0.024, Fig. 3A), using linear regression controlling for the time difference between age of onset and the tau-PET acquisition date, sex, education and diagnosis. In addition, we tested whether the epicenters of tau pathology (i.e., 10% of brain regions with highest baseline tau-PET positivity,) were more likely to be located in more globally connected regions in younger symptomatic AD patients (see Supplementary Fig. 1 for a mapping of epicenters; anatomical locations of the epicenters are labeled in the source data file). Supporting this, we found that younger age was indeed associated with stronger hub-ness (i.e., shorter connectivity-based distance to the rest of the brain) of tau epicenters in patients with symptomatic AD (ADNI: β = 0.300, p = 0 < 0.001, Fig. 2C, right panel; BioFINDER: β = 0.646, p < 0.001, Fig. 2D, right panel), using linear regression controlling for sex, education and diagnosis. Again, this result pattern was not observed in preclinical AD (ADNI: β = −0.028, p = 0.785, Fig. 2C, left panel; BioFINDER: β = 0.022, p = 0.943, Fig. 2D, left panel), controlling for sex and education. In ADNI, we repeated these analyses with informant-based ages of cognitive symptom onset. Here, we found congruent results, showing that younger age of onset was associated with stronger hub-ness of tau epicenters (i.e., shorter connectivity-based distance to the rest of the brain), controlling for the time difference between age of onset and the tau-PET acquisition date, sex, education and diagnosis (β = 0.352, p = 0.002, Fig. 3B). Together, these results support the hypothesis that younger AD symptom onset is associated with stronger tau pathology in functional hub regions that are strongly interconnected with the rest of the brain.
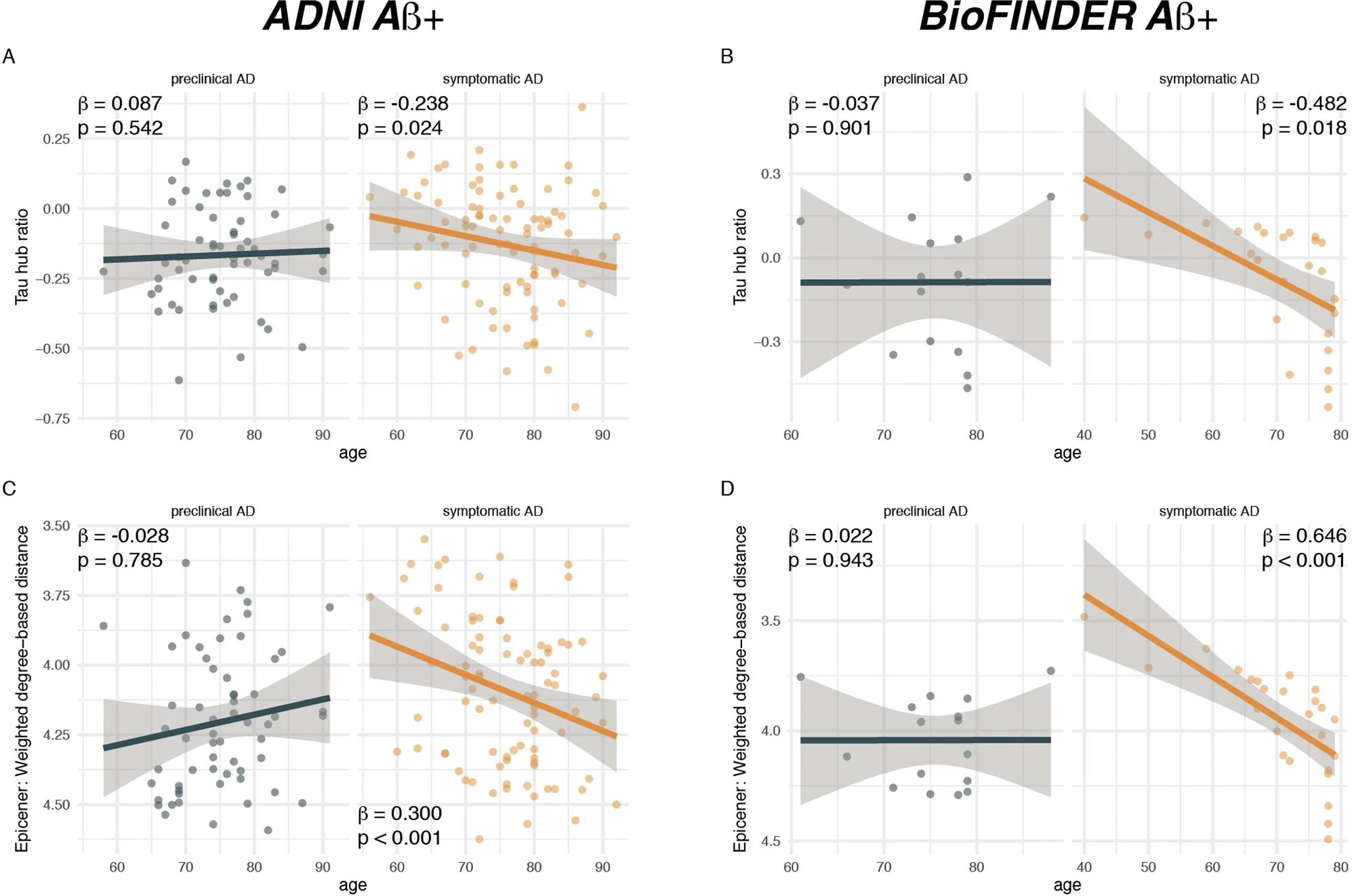
Younger age is associated with a higher hub ratio in symptomatic AD.
Scatterplots illustrating the association between age and the tau hub ratio for Aβ+ subjects of the A ADNI (N = 147) and B BioFINDER sample (N = 41), stratified by clinical status. C, D illustrate the association between age and the average weighted degree-based distance (i.e., shorter distance = higher connectivity) of tau epicenters (i.e., 10% of brain regions with highest tau-PET positivity which were determined on the subject level) in C ADNI and DBioFINDER. Note that the y-axis in C, D has been inverted, since lower values mean reflect higher hub-ness. β-values reflect standardized regression weights. All β- and two-sided p values were derived from linear regression controlling for sex, education (and diagnosis in symptomatic AD groups). Linear model fits (i.e., least squares line) are indicated together with 95% confidence intervals. Source data are provided as a Source Data file.
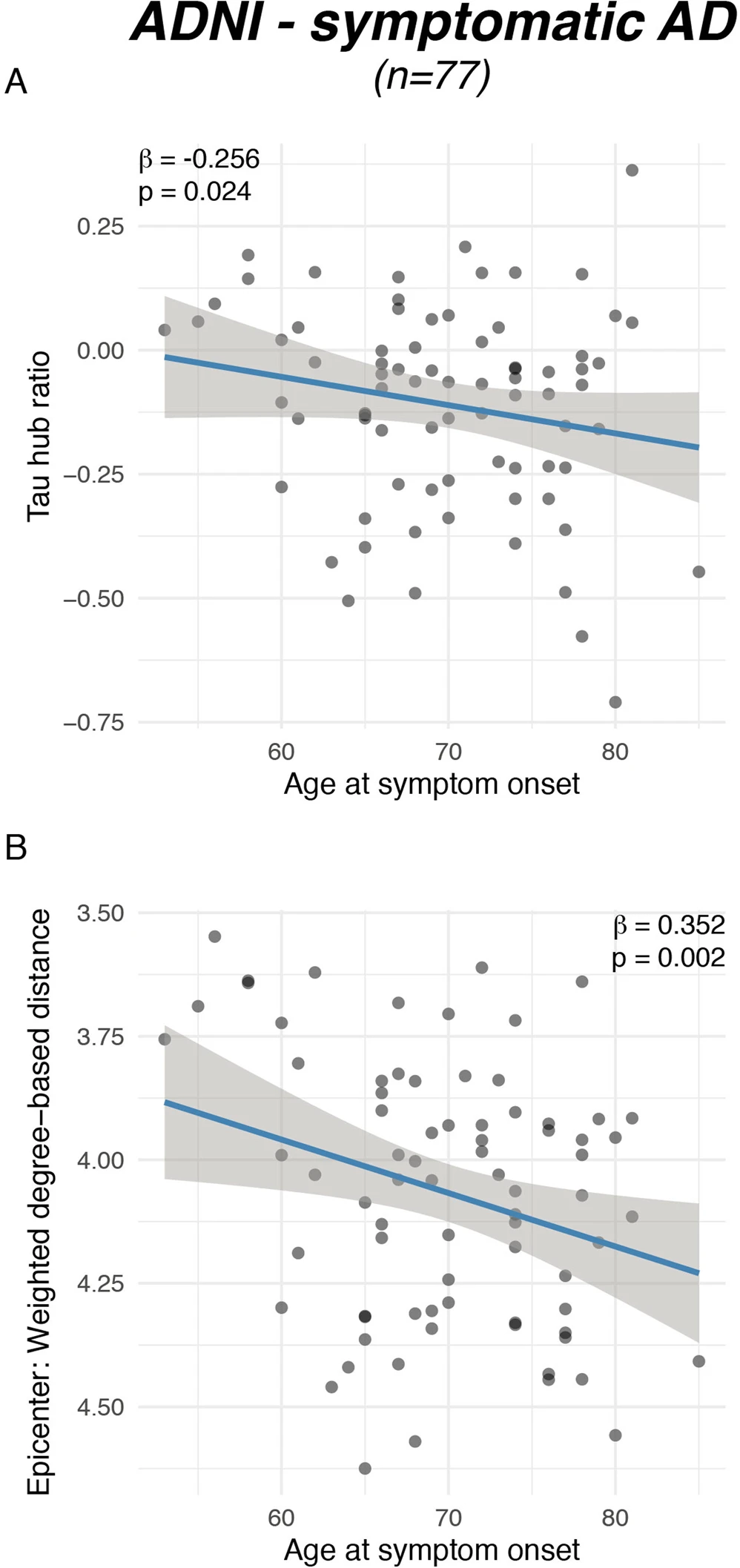
Younger symptom onset is associated with a higher tau hub ratio.
Scatterplots illustrating the association between (A) actual age of onset and the tau hub ratio for n = 77 symptomatic AD subjects of the ADNI sample as well as between (B) age at symptom onset and global connectivity-based distance of the tau epicenters. β-values reflect standardized regression weights. All β-and two-sided p values were derived from linear regression controlling for sex, education and diagnosis and the time difference between age of symptom onset and the first tau-PET scan. Linear model fits (i.e., least squares line) are indicated together with 95% confidence intervals. Source data are provided as a Source Data file.
In an exploratory analysis, we tested whether amyloid-positive ApoE4 non-carriers showed a stronger tau hub ratio, since previous work has suggested that ApoE4-carriage is associated with a more limbic-predominant pattern of tau pathology in AD patients16, whereas amyloid-positive ApoE4 non-carriers show a more neocortical tau pathology pattern42. Supporting this, we found that ApoE4-carriage was associated with a lower tau hub ratio in amyloid-positive (i.e., AD) patients (ADNI/BioFINDER: F = 4.816/4.404, Cohen’s d = 0.4/0.3, p = 0.036/0.038), while controlling for age, sex, education and diagnosis. All above-described results remained consistent when including hippocampal tau-PET in the analyses (see Supplementary Table 2).
Younger age and a higher tau hub ratio are associated with faster tau accumulation in symptomatic AD
In a next step, we tested whether younger age was associated with a faster tau accumulation rate (i.e., annual change rate in global tau positivity) in patients with symptomatic AD. In line with previous work3,4, we found that younger age was associated with a faster annual change rate in global tau-PET positivity in patients with symptomatic AD (ADNI: β = −0.284, p = 0.009, Fig. 4B, right panel; BioFINDER: β = −0.836, p < 0.001, Fig. 4C, right panel), using linear regression controlling for sex, education and diagnosis. As expected, no association between younger age and faster tau accumulation was detected in patients with preclinical AD (ADNI: β = −0.119, p = 0.389, Fig. 4B, left panel; BioFINDER: β = 0.208, p = 0.448, Fig. 4C, left panel), suggesting that faster tau accumulation or tau accumulation in general at younger age is specific for subjects with symptomatic AD. Next, we tested whether a higher tau hub ratio, indicating a more hub-like tau-PET deposition pattern, was associated with faster global tau accumulation. This analysis was motivated by our previous findings, showing that tau spreads from circumscribed epicenters to connected brain regions33,34. Thus, tau pathology in a globally connected hub region should result in more widespread tau spreading and faster tau accumulation. In contrast, tau in only weakly interconnected non-hub regions should result in slower spreading and tau accumulation (see Fig. 4A for an example illustration). Supporting this, we found that a higher tau hub ratio at baseline predicted faster subsequent tau accumulation in patients with symptomatic AD (ADNI: β = 0.347, p = 0.006, Fig. 4D, right panel; BioFINDER: β = 0.668, p = 0.002, Fig. 4E, right panel), controlling for sex, education and diagnosis. Exact tests using 1000 beta values from null-model tau hub ratios that were generated using shuffled connectomes as a reference confirmed these results (ADNI: p = 0.010; BioFINDER: p = 0.018). This result remained consistent when additionally controlling for age (ADNI: β = 0.259, p = 0.009; BioFINDER: β = 0.311, p = 0.026), suggesting a unique contribution of the tau hub ratio to faster tau accumulation. In patients with preclinical AD, we also found a significant association between a higher tau hub ratio at baseline and a faster subsequent tau accumulation rate in ADNI (β = 0.347, p = 0.006, Fig. 4D, left panel), controlling for sex and education, however this finding could not be replicated in BioFINDER (β = 0.199, p = 0.469, Fig. 4E, left panel). In summary, these results suggest that younger age and a more hub-like tau-PET retention pattern are associated with a faster accumulation of tau pathology in patients with symptomatic AD. Again, these results remained consistent when including hippocampal tau-PET (see Supplementary Table 2).
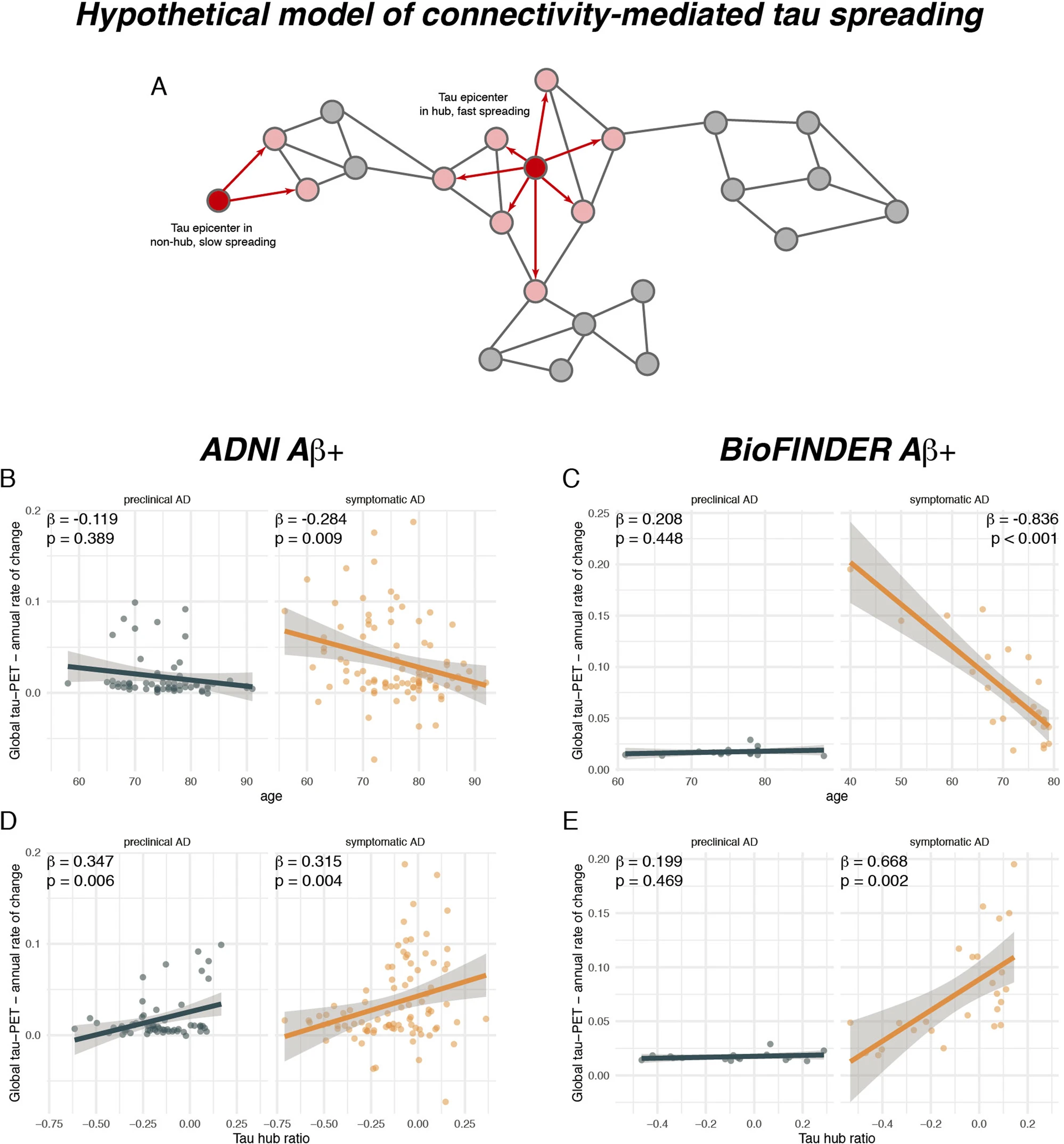
A higher tau hub ratio facilitates tau spreading.
A Schematic illustration of a hypothetical tau spreading model. When tau epicenters fall in a hub region with many connections to other regions, tau spread should be faster as compared to tau spread from an epicenter in a non-hub region with fewer connections. The association between age and annual tau-PET change rates for Aβ+ subjects is illustrated in B for the ADNI (N = 147) and C for the BioFINDER sample (N = 41), stratified by clinical status. Scatterplots showing the association the tau hub ratio and annual tau accumulation rates in D ADNI and E BioFINDER. β-values reflect standardized regression weights. All β-and two-sided p values were derived from linear regression controlling for sex, education (and diagnosis in symptomatic AD groups). Linear model fits (i.e., least squares line) are indicated together with 95% confidence intervals. Source data are provided as a Source Data file.
To further confirm that it is indeed the spatial distribution of tau pathology in hubs and not the overall severity of tau pathology that drives faster tau accumulation and earlier disease manifestation, we performed additional confirmatory analyses. Specifically, we conducted a sliding-window analysis from low to high global tau-levels analysis in Aβ+ subjects of ADNI and BioFINDER, and assessed for each window the mean annual tau-PET change and age, stratified by a high vs. low tau hub ratio (i.e., median split) or by high vs. low epicenter connectivity-based distance. We found significant interactions of global tau-PET and the tau hub ratio on the annual rate of tau accumulation (Supplementary Fig. 1A, B), where a higher tau hub ratio was associated with faster tau accumulation at higher baseline tau-PET levels. Similarly, we found an interaction between global tau-PET and the tau hub ratio on age in symptomatic AD patients, where a higher tau hub ratio was associated with younger age at higher tau-PET levels. These analyses support the view that a higher tau hub ratio is associated with faster tau accumulation at a given level of tau pathology in AD, as well as with younger age in symptomatic AD. Similarly, we found that at a given level of global tau-PET, more globally connected epicenters were associated with faster subsequent tau accumulation. Analyses are summarized in Supplementary Fig. 2A–F.
A higher tau hub ratio mediates the association between younger age and faster tau accumulation in symptomatic AD
Lastly, we assessed in symptomatic AD patients whether the effect of younger age on faster tau accumulation rates was mediated via stronger tau pathology in hub regions, i.e., a higher tau hub ratio. Using bootstrapped mediation analyses with 10,000 iterations, we found a significant mediation effect both in ADNI (β = −0.065, 95% CI [−0.153; 0.00], p = 0.039) and BioFINDER (β = −0.149, 95% CI [−0.371; 0.00], p = 0.046), controlling for sex, education and diagnosis. Mediation analyses are summarized in Fig. 5A, suggesting that a higher tau hub ratio mediates the association between younger age and faster tau accumulation rates in patients with symptomatic AD. These data remained consistent when also including hippocampal tau-PET (ADNI: β = −0.069, 95% CI [−0.164; −0.01], p = 0.024; BioFINDER: β = −0.149, 95% CI [−0.366; −0.01], p = 0.033). Using longitudinal cognitive data available in symptomatic AD patients of the ADNI sample, we could further show that faster annual tau accumulation was associated with a faster decline in a memory composite (i.e., ADNI-MEM, β = −0.322, p = 0.004, Fig. 5B, controlling for age, sex and education), suggesting that faster tau hub ratio-dependent accumulation of tau pathology may drive faster clinical deterioration in younger patients with clinical AD. Again, this association remained consistent when including hippocampal tau-PET (β = −0.315, p = 0.004). Confirming these analyses using the sliding-window approach introduced above, we found an interaction between baseline global tau-PET and the tau hub ratio on annual changes in ADNI-MEM, where a higher tau hub ratio was associated with faster cognitive decline at higher overall tau levels.
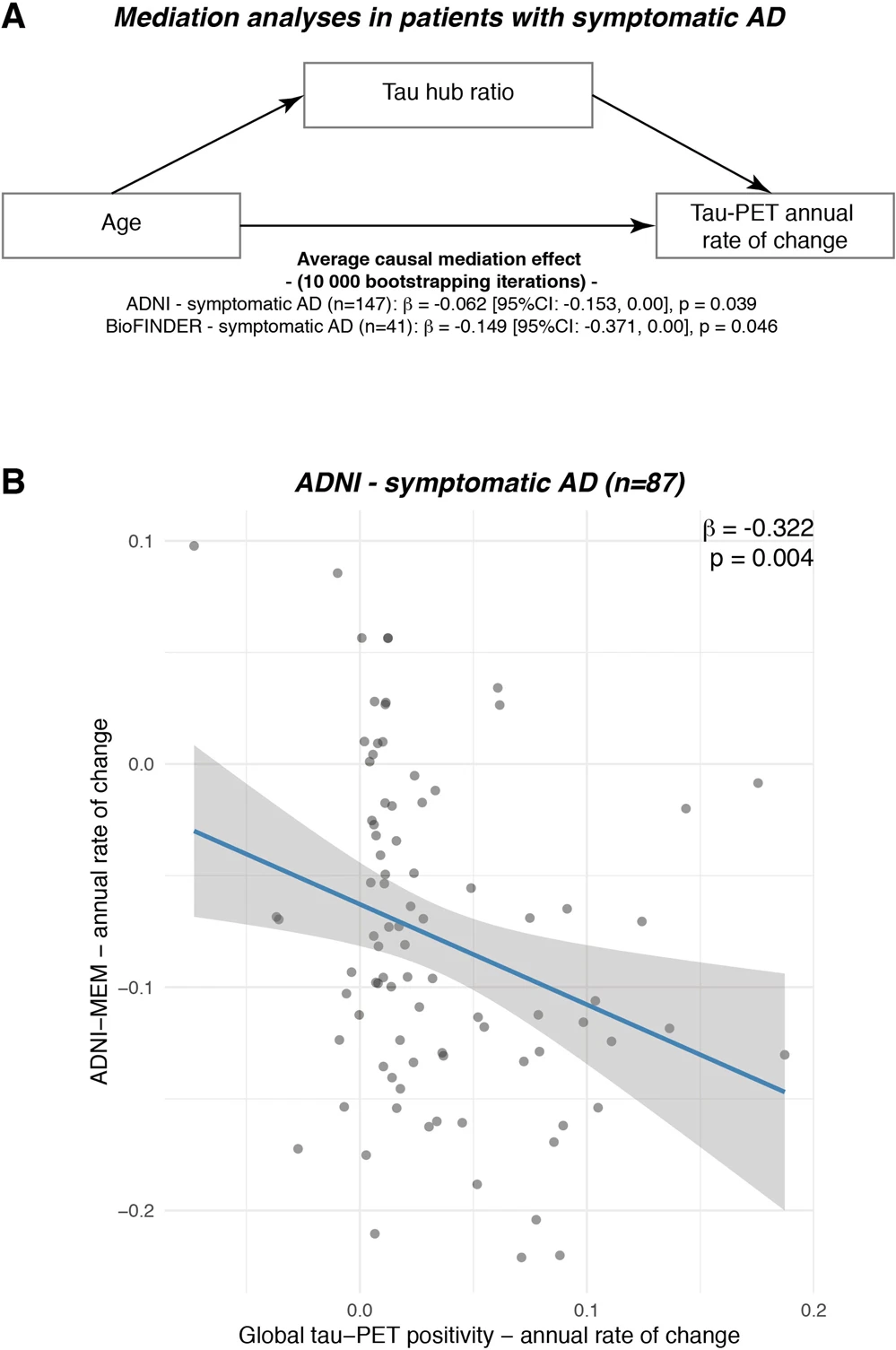
The association between younger age and faster tau accumulation is mediated by a higher tau hub ratio.
A Mediation analysis in patients with symptomatic AD (ADNI: N = 87, BioFINDER: N = 25), showing that the association between younger age and faster annual tau accumulation rates is mediated by tau hub ratio. Mediation effects (i.e., β-values, 95% confidence intervals [CI] and uncorrected two-sided pvalues) were determined based on bootstrapping with 10,000 iterations, while all paths were controlled for age, sex and education. B In the ADNI symptomatic AD sample with available longitudinal cognition on a memory composite (i.e., ADNI-MEM, n = 87), we plotted the association between tau-PET accumulation rates (x-axis) and cognitive change rates (y-axis). β-values reflect standardized regression weights. β-and two-sided p values were derived from linear regression, controlling for age, sex and education. Linear model fits (i.e., least squares line) are indicated together with 95% confidence intervals. Source data are provided as a Source Data file.
Discussion
The main findings of the current study were first, that younger patients with symptomatic AD show a more hub-like pattern of PET-assessed tau deposition, i.e., stronger tau in highly connected brain regions which are critical for domain-general cognition22 and which we have previously shown to be central for maintaining cognitive performance in AD25,26,27,43. Importantly, these associations were not detected for amyloid-positive, yet asymptomatic individuals (i.e., preclinical AD), in whom tau pathology was mostly absent. This supports previous findings of tau-pathology being a key driver of symptomatic disease manifestation in AD1,14,15,44, while adding important new evidence that stronger tau deposition in globally connected hubs is associated with earlier AD symptom manifestation. Second, we found that younger age and a stronger load of tau pathology in globally connected hubs predicted faster subsequent tau accumulation rates in symptomatic AD patients which was associated with faster decline in memory performance. Connectivity is assumed to be a key mediator of tau spreading31,33,34,35, hence these results favor the hypothesis that tau pathology in globally connected hubs early in the disease course may lead to more widespread propagation of tau pathology across connected brain regions, thereby driving faster cognitive deterioration in patients with earlier symptomatic AD manifestation14,15,30. To integrate our findings on how age, tau deposition patterns and tau accumulation rates are inter-related, we show that the association between younger age and faster tau accumulation in symptomatic AD patients is mediated by stronger tau pathology in hub regions. Together, the current independently validated findings provide key evidence that a predilection of tau pathology deposition toward globally connected brain hubs that are crucial for cognition22 may determine earlier symptomatic disease manifestation and accelerate connectivity-mediated tau spreading.
For our first finding, we show that patients with younger symptomatic manifestation of AD show stronger tau pathology in globally connected hubs in the fronto-parietal association cortex compared to inferior temporal non-hubs (Fig. 2). This finding aligns well with previous post-mortem10 and in vivo tau-PET evidence12,16,17,18,19, showing that younger symptomatic AD onset is associated with a more neocortical limbic sparing tau pathology pattern, whereas older age of symptom onset is associated with a limbic-predominant tau pathology pattern10. Similarly, previous studies found that younger symptomatic AD onset is associated with stronger fronto-parietal gray-matter atrophy5, hypoperfusion and glucose hypometabolism45,46, which have been shown to be closely associated with tau pathology in AD13. Together, this suggests that the spatial pattern rather than the mere extent of tau pathology may determine the likelihood, type and aggressiveness of symptom manifestation in AD1,12,47. The current findings critically extend these previous results by showing that younger symptomatic AD manifestation is specifically associated with a tau pathology pattern that resembles hub regions that are critical for domain general cognitive functioning22,40. Supporting a role of hubs for cognition in AD, we found previously that higher global connectivity fronto-parietal control network hubs such as the left frontal cortex is associated with a more efficient (i.e., small-world) global brain network topology48 and attenuated effects of posterior-cingulate glucose hypometabolism25, hippocampal atrophy43,49,50 or entorhinal tau pathology on cognitive performance27. This suggests that maintaining the integrity of hub regions may promote resilience to the impact of AD pathology on cognition49. In turn, damage to hub regions has been shown to diminish the efficient communication between brain networks51, and clinical studies in patients with cerebrovascular disease have shown that focal lesions in hubs cause brain-wide network disruptions52,53 and stronger impairment than lesions in non-hub regions54. Tau pathology has been shown to disrupt neuronal activity and connectivity in both preclinical29 and clinical studies55,56,57,58, hence early occurrence of tau pathology in hub regions may lead to an earlier impairment of global brain network function51 and thus earlier onset of cognitive impairment. Here, it will be an important next step to address whether higher tau pathology in hubs in fact drives a disruption of hub connectivity and thus an earlier symptom onset.
For our second finding, we could show that younger age and a stronger tau hub ratio are associated with faster tau accumulation in symptomatic AD. This is a critical extension of previous preclinical and clinical work, showing that brain connectivity mediates tau spreading33,34,35,59. Specifically, studies in mice and neuronal cell-cultures found that tau spreads trans-synaptically, where higher synaptic activity facilitates tau spreading31,32,59. Similarly, we and others reported in combined MRI and tau-PET studies in AD patients that seed-based connectivity of tau harboring epicenters predict tau spreading patterns, where tau spreads preferentially from epicenters to connected regions2,33,34,35,36,37. Thus, a tau harboring hub region with widespread connections may potentiate tau spreading to connected regions compared to a non-hub with fewer connections (i.e., as illustrated in Fig. 4A). Supporting this, we found that younger symptomatic AD patients had regions of highest tau pathology (i.e., epicenters) in more globally connected brain regions and that a stronger tau hub ratio was associated with faster tau accumulation. Importantly, our findings also offer a mechanistic explanation for previous results on faster tau accumulation rates in younger AD patients3,4, which is further supported by our mediation analyses showing that the association between younger age and faster tau accumulation rates is mediated by a stronger tau hub ratio in symptomatic AD. Importantly, faster tau accumulation was associated with faster decline in memory performance, supporting the view that faster tau accumulation drives the rapid clinical progression in AD patients with earlier symptom manifestation6,7,8,30. Yet, we caution that these results on cognition were restricted to ADNI and warrant further validation in future studies. Nevertheless, our results suggest that hubs may play an important role as distributors of tau pathology, driving more widespread tau propagation and thus clinical deterioration. Of note, we also found an association between the tau hub ratio and faster tau accumulation in preclinical AD patients in ADNI but not in BioFINDER, potentially since preclinical AD patients in ADNI already show slightly more advanced tau pathology than in BioFINDER (see Table 1) and may therefore be at higher risk of subsequent tau spreading38.
For interpreting the results of the current study, several caveats should be taken into account. First, Flortaucipir tau-PET shows considerable off-target binding in subcortical regions including the hippocampus and basal ganglia60. To address this, we excluded regions that are known to be severely affected by unspecific binding for our main analyses (e.g., hippocampus, basal ganglia) and further minimized any influence of Flortaucipir off-target binding by transforming tau-PET SUVRs to tau positivities using a Gaussian mixture modeling approach that has been previously applied by amyloid- and tau-PET studies to separate target- from unspecific binding33,36,61. In an exploratory approach, we also included the hippocampus using a pre-established hippocampal mask41 that excludes the choroid plexus (i.e., the main source of off-target binding), yielding fully consistent results with our main analyses (see Supplementary Table 2). Still, it is possible that unspecific binding influences our results, hence our results await further replication using second-generation tau-PET data with a better off-target binding profile. Second, the current study included only include a small number of patients that would be clinically considered as early-onset AD (i.e., below the age of 65, ADNI/BioFINDER, n = 8/4). Therefore, the current study is mostly limited to the variability of tau deposition in the age range of typical late-onset AD. To specifically replicate the role of tau pathology in hubs in subjects with a clinical disease onset before the age of 65, we encourage future studies to assess whether our results can be replicated in dedicated early-onset AD datasets such as the LEADs cohort (https://clinicaltrials.gov/ct2/show/NCT03507257), once sufficient data become available. Third, the current study exclusively used weighted degree (i.e., global connectivity) as a measure of hub-ness62,63, which yielded similar hub patterns as in previous studies20,63. We specifically selected this measure, since it is relatively easy to interpret (i.e., strength of connectedness to the rest of the brain) and well-suited to test the effect of connectivity on tau spreading. We are, however, aware of the breadth of graph-theoretical measures that have been proposed to quantify the hub-ness of a given brain region, yet many of these measures are relatively abstract (e.g., betweenness-centrality, participation coefficient), and often highly intercorrelated21,64. To avoid introducing additional and potentially redundant hub measures to an already complex set of analyses, we thus refrained from repeating the analyses with alternative measures of hub-ness. Fourth, other modulating factors such as reserve and resilience may influence symptom onset and progression rates in AD65. To account for inter-individual differences in reserve and resilience, all analyses were statistically controlled for years of education, i.e., a well-established proxy of reserve and resilience in AD65. However, we cannot exclude that inter-individual differences in reserve/resilience may further modulate symptom onset and progression rates. Lastly, we would like to highlight that we did not perform partial volume correction since longitudinal T1-weighted MRI data was not consistently available and since previous studies showed that longitudinal tau-PET changes can also be reliably detected without partial-volume correction66,67.
In conclusion, our independently validated results provide novel evidence that younger symptomatic AD patients show stronger tau pathology in globally connected hubs, which may drive faster tau spreading and accelerated cognitive decline. This suggests that earlier symptom manifestation is not driven by specific pathophysiological characteristics11, but rather by a tau distribution pattern10,12 that preferentially targets brain hubs important for cognitive function22,25,26,27,49,50. These results converge well with previous findings, showing that heterogeneous tau distribution patterns are associated with heterogeneous clinical trajectories, variable symptom onset and disease progression1,2,10,12,14,15,33,47. Here, it will be a key future goal to identify potential determinants of spatially variable tau pathology onset, such as differential gene expression patterns68,69,70, pre-existing tau pathology (e.g., related to traumatic brain injury)71,72, or premorbid differences and/or heterogeneity in brain network architecture73. Knowledge about drivers of tau onset, heterogeneous tau spreading patterns and clinical trajectories may become important to facilitate precision-medicine prediction of cognitive and pathological progression, as well as for patient stratification in clinical trials14,74.