
CaV1.2 channelopathic mutations evoke diverse pathophysiological mechanisms

By Moradeke A. Bamgboye, Kevin G. Herold, Daiana C.O. Vieira, Maria K. Traficante, Philippa J. Rogers, Manu Ben-Johny, and Ivy E. Dick
Excerpt from the article published in BioRxiv 2022.06.13.495975; Posted June 16, 2022, DOI: https://doi.org/10.1101/2022.06.13.495975
Editor’s Highlights
- CaV1.2 L-type Ca2+ channels are perhaps the most prevalent of the voltage-gated Ca2+channels, existing in cardiac, neuronal, and smooth muscle cells.
- Mutations in CaV1.2 have been linked to severe cardiac and neurological disorders which are often fatal in early childhood.
- The first pathogenic mutation in CaV1.2 was identified in 2004 and was shown to cause a severe multisystem disorder known as Timothy syndrome (TS) with significant neurodevelopmental deficits including autism spectrum disorder (ASD).
- While pathogenic mutations in CaV1.2 have been identified across multiple distinct channel regions, many appear to cluster near a distal S6 and exert their impact through a change in channel gating.
- Neuronal effects of some CaV1.2 mutations may be independent of Ca2+ permeation.
- Two mutations I1166V and I1166T cause a marked change in single-channel properties. Mutations E1496K and I1475M produce a left shift in channel activation
Abstract
The first pathogenic mutation in CaV1.2 was identified in 2004 and was shown to cause a severe multisystem disorder known as Timothy syndrome (TS). The mutation was localized to the distal S6 region of the channel, a region known to play a major role in channel activation. TS patients suffer from life-threatening cardiac symptoms as well as significant neurodevelopmental deficits including autism spectrum disorder (ASD). Since this discovery, the number and variety of mutations identified in CaV1.2 has grown tremendously, and the distal S6 regions remains a frequent locus for many of these mutations. While the majority of patients harboring these mutations exhibit cardiac symptoms which can be well explained by known pathogenic mechanisms, the same cannot be said for the ASD or neurodevelopmental phenotypes seen in some patients, indicating a gap in our understanding of the pathogenesis of CaV1.2 channelopathies. Here, we use of whole cell patch clamp, quantitative Ca2+ imaging, and single channel recordings to expand the known mechanisms underlying the pathogenesis of CaV1.2 channelopathies. Specifically, we find that mutations within the S6 region can exert independent and separable effects on activation, voltage-dependent inactivation (VDI) and Ca2+-dependent inactivation (CDI). Moreover, the mechanisms underlying the CDI effects of these mutations are varied and include altered channel opening and possible disruption of CDI transduction. Overall, these results provide a structure-function framework to conceptualize the role of S6 mutations in pathophysiology and offer insight into the biophysical defects associated with distinct clinical manifestations.
Introduction
CaV1.2 L-type Ca2+ channels are perhaps the most prevalent of the voltage-gated Ca2+channels, existing in cardiac, neuronal and smooth muscle cells (1). Ca2+ entry through these channels is precisely controlled through multiple forms of regulation including voltage-dependent activation, voltage-dependent inactivation (VDI) and Ca2+-dependent inactivation (CDI) (2–6). Each of these regulatory processes are vital, and disruption of them is expected to produce adverse physiological consequences. Timothy syndrome (TS) represents one such class of mutations, in which a single point mutation within CaV1.2 leads to a severe multisystem disorder characterized by developmental delays, autism spectrum disorder (ASD) and profound long-QT syndrome (LQTS) (7–9). The underlying cause of TS was first described in 2004 as a single point mutation (G406R) within the distal IS6 region of CaV1.2 (8). This form of LQTS is among the most severe presentation of this syndrome, with patients exhibiting QT intervals between 480-730 ms (7, 8, 10, 11). As a result, patients suffer from life threatening arrhythmias, which are often fatal in early childhood. Furthermore, TS represents one of the most penetrant monogenic forms of ASD (7, 8, 12). Since the first discovery of the G406R mutation, a growing number of mutations have been identified in CaV1.2, most of which result in significant cardiac phenotypes (7, 8, 13–15). However, only select mutations also cause neurological phenotypes including ASD or developmental delay (7, 8, 13, 15, 16). As the number and variety of identified CaV1.2 mutations continues to expand, so to does the phenotypic variation observed in patients. In the heart, the importance of CaV1.2 inactivation has long been recognized and there is a clear mechanistic link between excess Ca2+ entry through CaV1.2 and prolongation of the cardiac action potential (AP) (17–19), the cellular hallmark for LQTS. However, the mechanistic link between CaV1.2 mutations and neurological effects remains much less clear. Mutations within both CaV1.2 and CaV1.3 channels which cause large effects on channel gating have been linked to ASD, however some CaV1.2 mutations with significant effects on channel gating produce no neurological phenotype, and simple gain-of-function/loss-of-function descriptions of CaV1.2 mutations result in little correlation with patient phenotypes (13, 20, 21).
The original G406R TS mutation is located within the distal IS6 region of the channel (8), as is the G402S mutation which was identified soon after (7). Both mutations produce a marked deficit in VDI (7, 22), fitting with prior work in the CaV1.3 channel which demonstrated that residues within the distal S6 region may be critical to the function of a ‘hinge-lid’ inactivation scheme (23). Moreover, the S6 is known to play a critical role in channel activation (23–26). It is therefore not surprising that each of these mutations alters the voltage dependence of channel activation, although in opposing directions (G406R, hyperpolarizing shift; G402S, depolarizing shift) (22). Finally, both mutations have been shown to cause a decrease in CDI, demonstrating multiple effects on channel gating. Interestingly, the effect on CDI for many mutations within this region of both CaV1.2 (including G402S and G406R) and CaV1.3 has been demonstrated to be a direct result of the change in channel activation (22, 23), implying that the S6 region of the channel may not play a direct role in CDI. While a growing number of CaV1.2 mutations have been identified across multiple channel regions, a large subset of these pathogenic mutations appear within distal S6 regions of the channel. Thus, there may be common mechanistic elements which can be elucidated by evaluating the effects of these S6 mutations.
Here, we evaluate the biophysical impact of select CaV1.2 mutations on CDI, VDI and channel activation, focusing primarily on mutations near the distal S6 region of the channel. Despite the locus of these mutations near the channel activation gate, we find that not all S6 mutations exert an effect on channel activation. In contrast to the canonical TS mutations, we find that the S6 mutations within this study have more selective effects on channel properties, sometimes altering CDI or VDI independently. Further investigation into the mechanisms underlying the gating changes in CDI-altering mutations reveals a remarkably diverse impact of S6 mutations. In particular, two mutations (I1166V and I1166T) caused a marked change in single channel properties indicative of enhanced entry into mode 2 gating, while another distal S6 mutation (I1475M) resulted in a selective CDI deficit, implicating a greater role for the IVS6 region in CDI. Finally, we show that these biophysical changes produce a difference in the response of the channels to a neurological stimulus, demonstrating that the neurological phenotype may be most strongly associated with enhanced channel activation.
Results
Pathogenic CaV1.2 mutations often appear near the distal S6
The first TS mutations described (G406R and G402S) were both located within the IS6 region and caused changes to channel activation (7, 8, 22). Since this first description, numerous additional mutations in proximity of the distal S6 of Cav1.2 have been reported (13, 14, 16, 27). Given the importance of this region in channel regulation, we chose several pathogenic mutations near a distal S6 region of the channel to focus on. Fig. 1A displays the locus of each mutation on a cartoon of the CaV1.2 channel, with the S6 regions highlighted in blue for easy reference. Mutation L762F (14) resides just past the IIS6 region and has been shown to cause a cardiac selective phenotype. We next considered two mutations within the IIIS6 region. Interestingly I1166T and I1166V (13) occur at the identical residue, yet produce distinct phenotypes, with I1166T producing a multisystem disorder including neurological effects, while I1166V causes a cardiac selective effect. From the IVS6 domain we focused on the mutation I1475M, which has been described as cardiac selective in patients (13). Finally, we considered the effect of one mutation which resides outside an S6 region. E1496K resides within the EF-hand located on the C-tail of the channel and is associated with a cardiac-selective phenotype (13). This mutation is included in this analysis as this region has previously been identified as critical for VDI (26, 28), and has been suggested to cause a shift in channel activation (13) similar to the effects seen with S6 mutations.
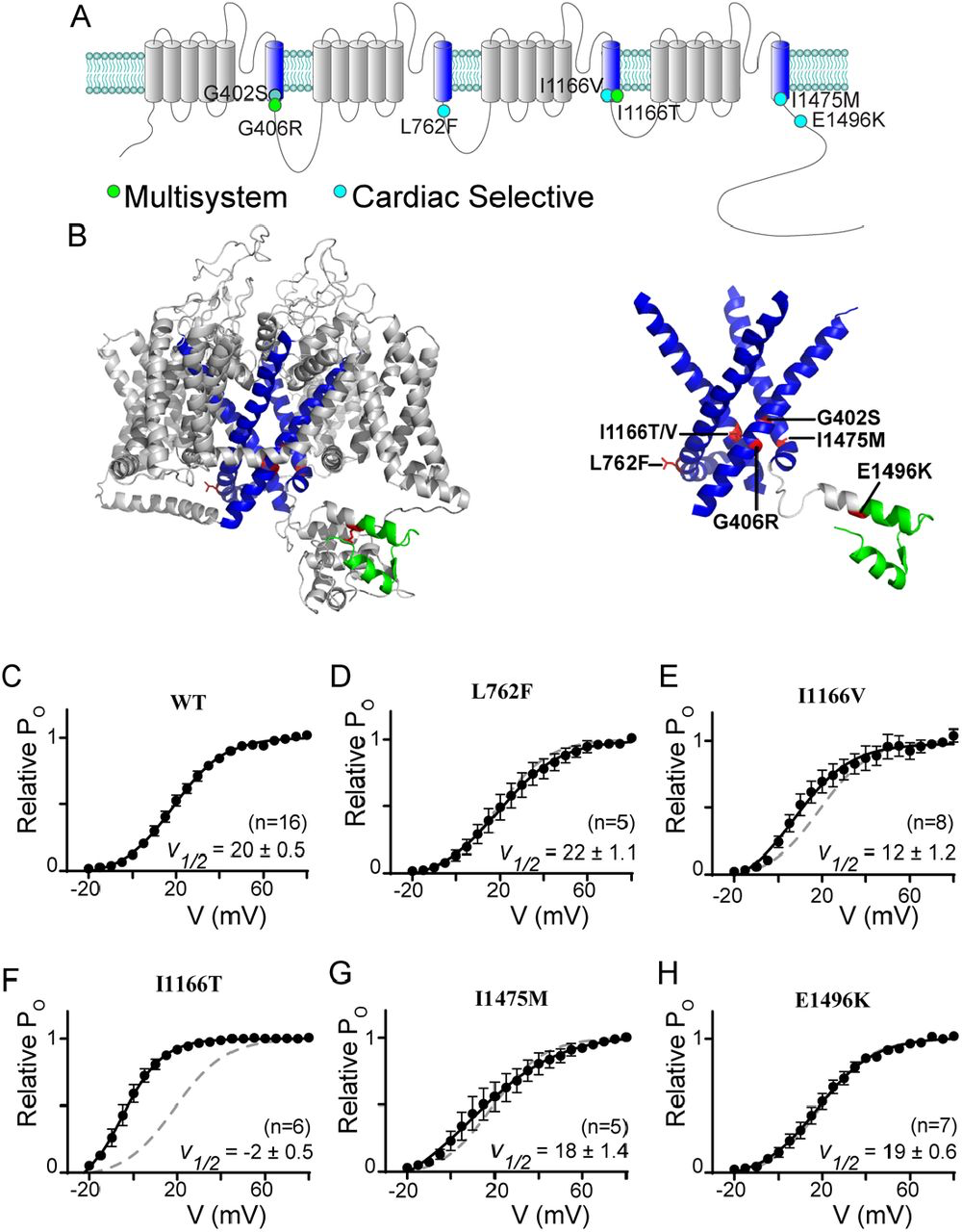
CaV1.2 mutations often reside near the distal S6 region
(A) Cartoon depicting the location of the canonical TS mutations along with the mutations described in this study. The S6 helices are in blue; the mutations associated with a multisystem disorder including neurological symptoms are in green; and the cardiac selective mutations are in cyan. Note that the G402S mutation had been associated with a multisystem disorder, but the neurological effects have been attributed to hypoxic injury secondary to the cardiac effects (54), we therefore shade this mutation to note the distinction. (B) Homology model of CaV1.2 demonstrating the locus of the mutations relative to the S6 helices shown in blue. Mutations are highlighted in red, EF-hand in green. Right: S1-5 helices are removed from the structural model to facilitate visualization of the mutations near the activation gate of the channel. (C) Activation curves determined by a standard tail current protocol (22). For all mutants WT data is reproduced as the dashed curve. Data is plotted ± SEM. V½ values were determined by a Boltzmann fit and are displayed ± standard error.
To fully appreciate the location of the mutations on the channel, we generated a homology model of CaV1.2 based on the structure of CaV1.1 (29, 30), enabling visualization of the mutation loci and surrounding residues (Fig 1b). The S6 regions are highlighted in blue, and visualization of the model structure with only the S6 regions displayed clearly demonstrates the location of the mutations near the helical-bundle that forms the channel activation gate, with the exception of the E1496K mutation which resides within the EF hand highlighted in green. Interestingly, while mutation L762F is predicted to lie just distal to the IIIS6 based on sequence, the homology model demonstrates that it does indeed reside on the S6 helix, which is extended in this domain.
Given the location of these mutations, we expect many may have effects on channel activation, as we previously showed for G406R and G402S (22). We therefore utilized a tail-current protocol (22) to define the relative activation curve for each mutation (Fig. 1C-H). Interestingly, despite its locus on the IIS6 helix, L762F did not significantly change the V1/2 of activation (Fig. 1D). However, when we explored the effect of I1166V, we found a small left shift in channel activation. Evaluation of I1166T revealed a much larger left shift, demonstrating the critical nature of this locus in channel activation. Next, we measured the effects of I1475M and E1496K and found that neither caused a change in the V1/2 of activation. Thus, only two of the S6 mutations investigated directly impact channel activation.
CaV1.2 mutations have selective effects on channel inactivation
Both canonical TS mutations have significant effects on channel inactivation, nearly eliminating VDI and blunting CDI (22). These effects have each been shown to have a clear impact on the cardiac action potential consistent with the LQTS phenotype of the patients (19, 22, 31). We therefore considered the impact of each of the mutations in this study on VDI and CDI. We begin by looking at VDI, which can be quantified through the use of a beta subunit permissive of VDI, such as β1b (32), with Ba2+ as the charge carrier so as to remove the confounding effect of Ca2+ dependent processes such as CDI. Under these conditions, VDI can be viewed as the decay in current during a 300 ms step depolarization (Fig. 2A, blue shade). Quantification of VDI is given by the metric r300, which is the ratio of current remaining after 300 ms as compared to peak current such that a value of 1 would equate to no VDI, while 0 would indicate complete inactivation. Plotting the r300 for WT CaV1.2 demonstrates the expected increase in VDI as a function of voltage (Fig. 2A, right). We next considered whether a VDI deficit may underlie the phenotype of patients harboring one of the mutations which lacked an activation shift. Indeed, evaluation of L762F demonstrated a near complete loss of VDI (Fig. 2B). Looking at E1496K (Fig. 2C), we saw a significant decrease in VDI, although to a lesser extent as compared to L762F. However, when we evaluated I1475M we found that VDI was preserved, leaving the mechanism underlying the patient phenotype for this mutation unresolved. Finally, we evaluated the mutations I1166V and I1166T, which altered channel activation; however, VDI remained unperturbed compared to WT.
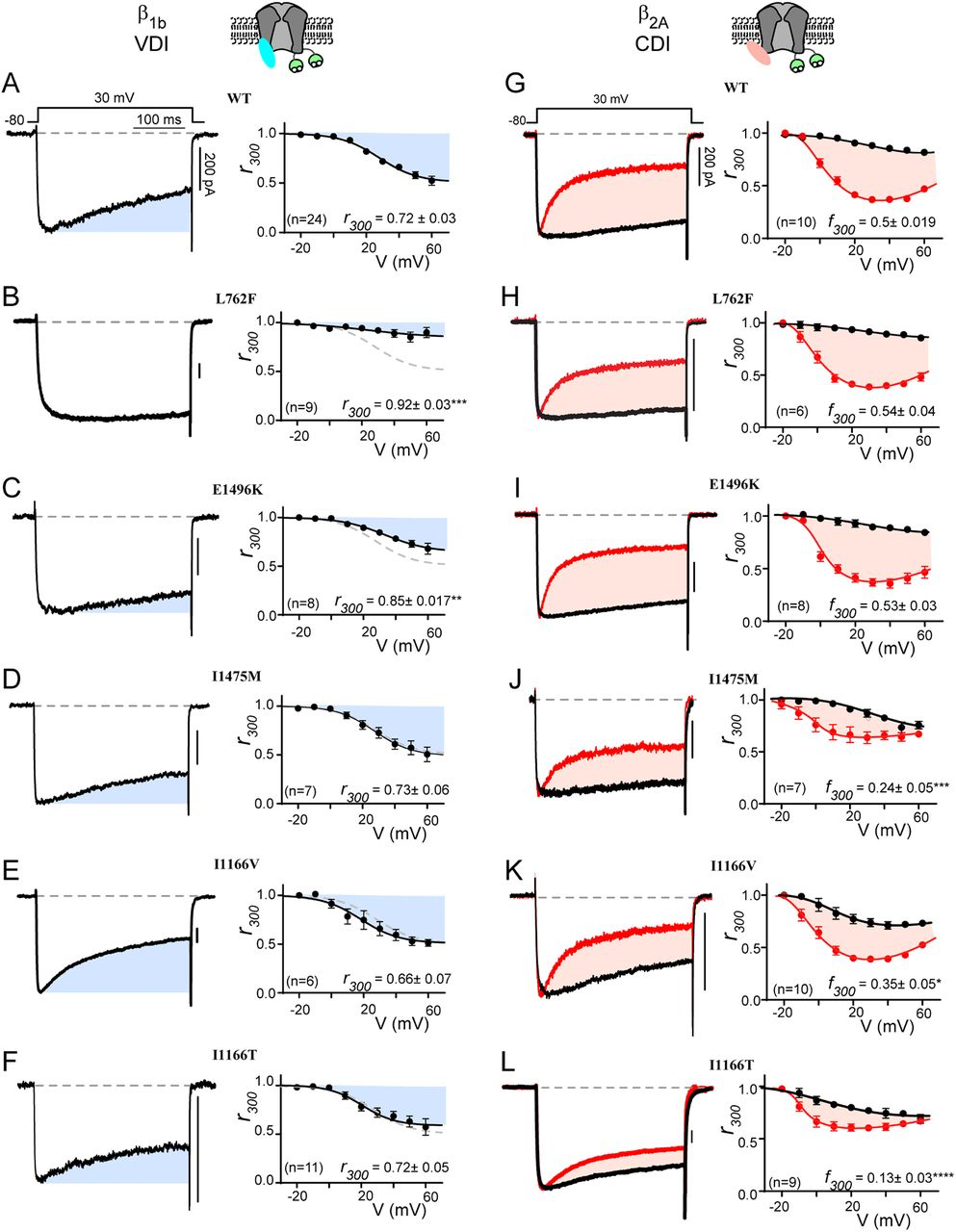
CaV1.2 mutations selectively alter CDI vs. VDI
(A) Exemplar whole cell current traces (left) in Ba2+ demonstrate robust VDI (shaded blue) in WT CaV1.2 expressed with the β1b auxiliary subunit. Population data plotted for multiple voltages (right) demonstrates an increase in VDI with increasing voltage. Blue shading indicates the amount of VDI. r300 quantifies VDI as the ratio of current remaining after 300 ms and is displayed for the 30 mV step. Data is plotted ± SEM here and throughout. (B, C) VDI is significantly decreased by mutations L762F and E1496K (D-F) Mutations I1475M, I1166V and I1166T (respectively) do not alter VDI. (G) Exemplar whole cell current traces (left) in Ba2+ (black) and Ca2+ (red) demonstrate robust CDI (shaded peach) in WT CaV1.2 expressed with the β2A auxiliary subunit. Population data plotting the r300 value for multiple voltages (right) demonstrates minimal VDI (black) under these experimental conditions, and an expected U-shaped dependence of CDI on voltage (red). Peach shading indicates the amount of CDI, quantified by the difference between r300 values in Ca2+ vs. Ba2+ (f300). The f300 value is given for the 30 mV data and is displayed ± SEM. Error bars indicate SEM here and throughout. (H, I) CDI is not significantly altered by mutations L762F or E1496K. (J-L) Mutations I1475M, I1166V and I1166T (respectively) reduce CDI, with I1166T having the largest effect. (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001, determined via a one-way ANOVA with Dunnett’s multiple comparison test)
We next considered the effect of each mutation on CDI. To do this, we utilized the β2A subunit which minimizes VDI (32), thus enhancing resolution of CDI in relative isolation. This decrease in VDI can be seen in the Ba2+ current trace (Fig. 2G, black), where inactivation is minimal. However, when Ca2+ is now used as the charge carrier through the channel (Fig. 2G, red), robust inactivation can be seen and CDI can be defined as the difference between the inactivation in Ca2+ as compared to Ba2+ (peach shaded area). Again, the metric r300quantifies the extent of inactivation in Ca2+ (red) and Ba2+ (black), while f300 is quantified as the difference between the Ca2+ and Ba2+ r300 values and provides a metric of pure CDI. Repeating this measurement for the L762F and E1496K mutations demonstrated no effect on CDI (Fig. 2H, I), consistent with the lack of change in channel activation. However, evaluation of I1475M, which also displayed normal activation parameters, revealed a significant decrease in CDI across multiple voltages (Fig. 2J). Thus, this mutation deviates from previous L-type channel mutations which caused a change in CDI secondary to an activation change (22, 23). Finally, I1166V and I1166T (Fig. 2K, L) also displayed a significant decrease in CDI, with I1166T again displaying a more severe effect. Overall, evaluation of these mutations demonstrates more selective effects on channel regulation, with separable effects on VDI, CDI and activation.
Reduced CDI at maximal Ca2+ concentrations revealed by Ca2+ uncaging
Our previous work has demonstrated that a reduction in CDI can occur through more than one mechanism (22). We therefore wanted to look more closely at the underlying deficits in the three CDI deficient channels. To do this, we considered an allosteric model for CaV1.2 gating in which channels initially open in a Ca2+ free configuration with a relatively high open probability (PO), known as mode 1 (33). Upon Ca2+ binding to calmodulin (CaM,) channels transition into a low PO state (mode Ca) (34), resulting in CDI. Within this scheme, CDI which is viewed at the whole cell level consists of two components: FCDI is the fraction of channels that enter mode Ca and is dependent on the Ca2+ driving force; while CDIMax is the maximum amount of CDI you would get if all channels were to enter mode Ca2+, and is dependent on the difference between the PO of mode 1 vs. mode Ca (Fig. 3A) (23). Our prior work has demonstrated that pathogenic CaV1.2 mutations can exert an effect on CDI by changing either of these two components (22). We therefore sought to measure the effect of the CDI-altering mutations on CDIMax. To do this, we utilized Ca2+ photo-uncaging paired with simultaneous patch clamp recordings and Ca2+ imaging. Within this setup, our goal was to drive CDI with a known concentration of Ca2+, enabling us to determine the extent of CDI as a function of Ca2+ concentration. DM-Nitrophen (DMNP) was used as the Ca2+ cage and added to our intracellular solution along with the Ca2+ indicator dye Flo4-FF. We could then release Ca2+ into the cytosol of the cell using a UV flash (Fig. 3A), while quantifying the amount of Ca2+ released through fluorescent imaging (22).
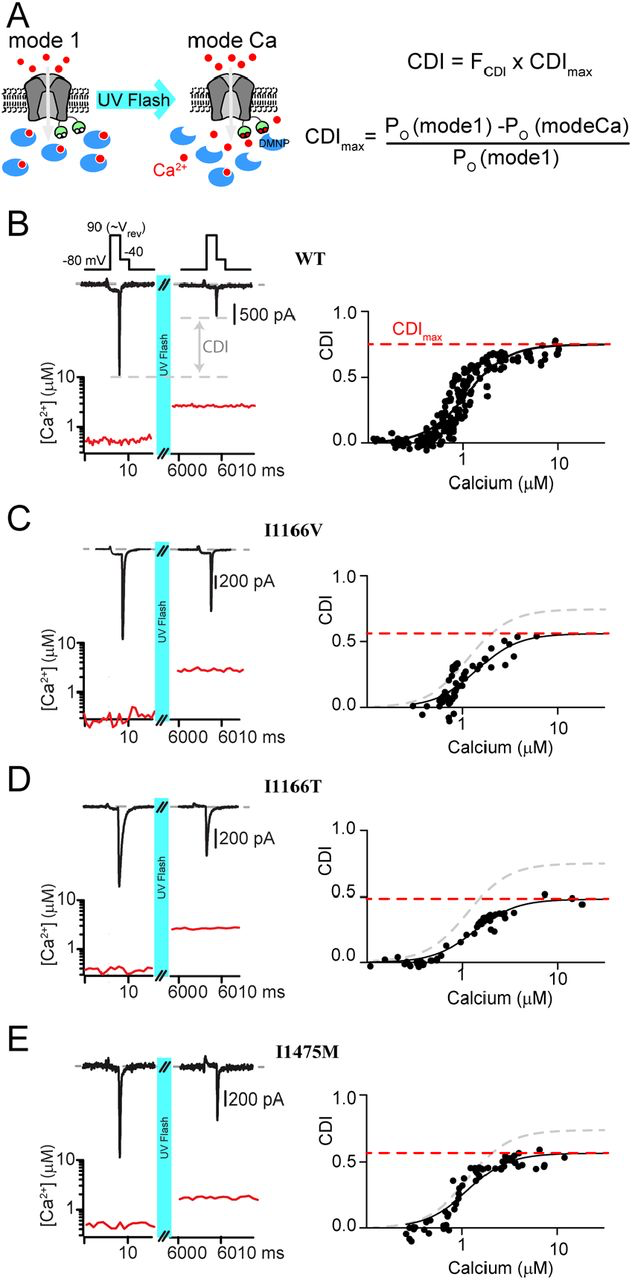
Measuring CDI as a function of Ca2+ concentration
(A) Left: Cartoon demonstrating the experimental setup. The Ca2+ cage DMNP (blue) is dialyzed into the cytosol through the patch pipette, keeping intracellular Ca2+ low at baseline, enabling mode 1. Application of a UV flash releases the Ca2+ cage, driving channels into mode Ca, which exhibits a reduced open probability. Right: Equations describing total CDI as a function of FCDI, the fraction of channels in mode C;, and CDIMax, the maximum amount of CDI that can be achieved if all channels were to enter mode Ca. (B) Top: Exemplar WT tail current data used to measure CDI in response to an increase in cytosolic Ca2+. Depolarization to the reversal potential enables channel opening without Ca2+ influx, followed by a step to the test potential of 40 mV, where a tail current is elicited. Tail current amplitude is significantly reduced following UV uncaging, indicative of CDI. Bottom: Ca2+ concentration is measured during the recording (red) using quantitative Ca2+ imaging. Right: Repetition of the protocol for many concentrations of Ca2+ resolves the CDI vs. Ca2+ relation. Data is fit by a Boltzmann distribution, with CDIMaxdisplayed as the dashed red line. (C-E) Application of the protocol to I1166V, I1166T and I1475M (respectively) demonstrates a decrease in CDIMax for all mutations, with III66T having the larges effect.
Our setup is similar to our prior study in which we characterized CaV1.2 CDI as a function of Ca2+ (22);, however, in that case we utilized a mutant CaV1.2 channel to prevent intracellular Ca2+ release from causing pore-block(35). However, the ability to quantify Ca2+ dependent regulation in WT Ca2+ channels has since been demonstrated for CaV2.1 channels (36). Given the potential for a channel mutation to impact our results, we opted to adapt this method for unaltered CaV1.2 channels. In order to do this, we utilized Ca2+ as the charge carrier, thus bypassing the pore block which would be induced following Ca2+ uncaging in the presence of an alternative charge carrier (22). Next, we ensured that the cytosolic Ca2+concentration would represent the Ca2+ concentration at the mouth of the channel by minimizing Ca2+ entry through the channel. Specifically, we applied a short step to the reversal potential of the channel (90 mV), which allowed channels to open without significant entry of Ca2+. Tail currents were then measured during a subsequent step to -40 mV; chosen so as to allow channels to fully close with kinetics enabling robust resolution of peak tail currents. A UV flash was then applied, elevating intracellular Ca2+, and the tail current protocol was repeated. Under these conditions, CDI was quantified as the difference in peak tail current following uncaging relative to current prior to Ca2+ release (Fig. 3B, left). Quantitative Ca2+ imaging then provided a readout of the cytosolic Ca2+ concentration for each tail current measured (Fig. 3B, red) and repeating the protocol for variable amounts of Ca2+ released enabled the generation of a relation between CDI and cytosolic Ca2+ (Fig. 3B, right). Importantly, repeated application of the tail current protocol produced stable current amplitudes and did not alter the measured cytosolic Ca2+ concentration. For WT channels, we obtained a CDI curve with a KD= 1.1 ± 0.04 µM, Hill coefficient = 2 and CDImax = 0.75 ± 0.02; a result nearly identical to our prior measurements with the mutant channel (22).
We next applied our Ca2+ uncaging protocol to the channels harboring CDI-altering mutations. Our previous work has demonstrated that a left shift in channel activation can be causative of a decrease in CDImax (22). We therefore began by considering whether the modest left shift in activation due to the I1166V mutation also caused a reduction in CDImax. Indeed, application of our tail current protocol to channels harboring the I1166V mutation (Fig. 3C) demonstrated a significant decrease in CDImax (KD = 1.4 ± 0.15, Hill coefficient = 2 and CDImax = 0.56 ± 0.05). We next considered the effect of the I1166T mutation, which produced a large left shift in channel activation. Likewise, this mutation resulted in a robust decrease in CDImax (KD = 1.27 ± 0.08, Hill coefficient = 2 and CDImax = 0.48 ± 0.02). Finally, we considered the effect of the I1475M mutation, which had no shift in channel activation. Despite this lack of activation change, we found that this mutation also caused a decrease in CDImax (KD = 1.1 ± 0.09, Hill coefficient = 2 and CDImax = 0.57 ± 0.02). Given the lack of shift in the activation curve for this mutation, this result indicates that the loss of CDI proceeds from a distinct mechanism not previously described for CaV1.2 mutations.
Single channel properties reveal a mode-switch in CaV1.2 mutant channels
All CDI-altering mutations in this study demonstrated a decrease in CDImax (Fig. 3), a metric dependent on the open probability of the channel (37). FCDI, on the other hand, cannot be directly measured but is determined by the Ca2+ entering the channel during each opening. It is therefore dependent on single-channel parameters including channel conductance and open probability. Therefore, to fully understand the mechanism underlying the CDI deficit we undertook single channel recordings for each CDI-altering mutation. For these experiments, Ba2+ was used as the charge carrier and channels were expressed with the β2A subunit in order to facilitate the evaluation of single channel properties in the absence of inactivation (32). Application of a voltage ramp across a single CaV1.2 channel resulted in channel openings with a conductance of 0.018 pA mV-1 (Fig. 4A), as previously recorded for this channel under these experimental conditions (22). Averaging numerous sweeps across multiple cells enabled the resolution of a PO curve (Fig. 4B, black) which could be well fit by a Boltzmann distribution (red). Moreover, we measured the individual open durations during the ramp and found that channels consistently exhibited brief openings, previously described as mode 1 gating (Fig. 4C) (33). After collecting hundreds of sweeps, we added Bay K 8644 which is known to induce mode 2 gating (Fig. 4A, gray box), in which channels open at more negative potentials and with significantly longer open durations. This maneuver ensures accuracy when counting the number of channels within each patch.
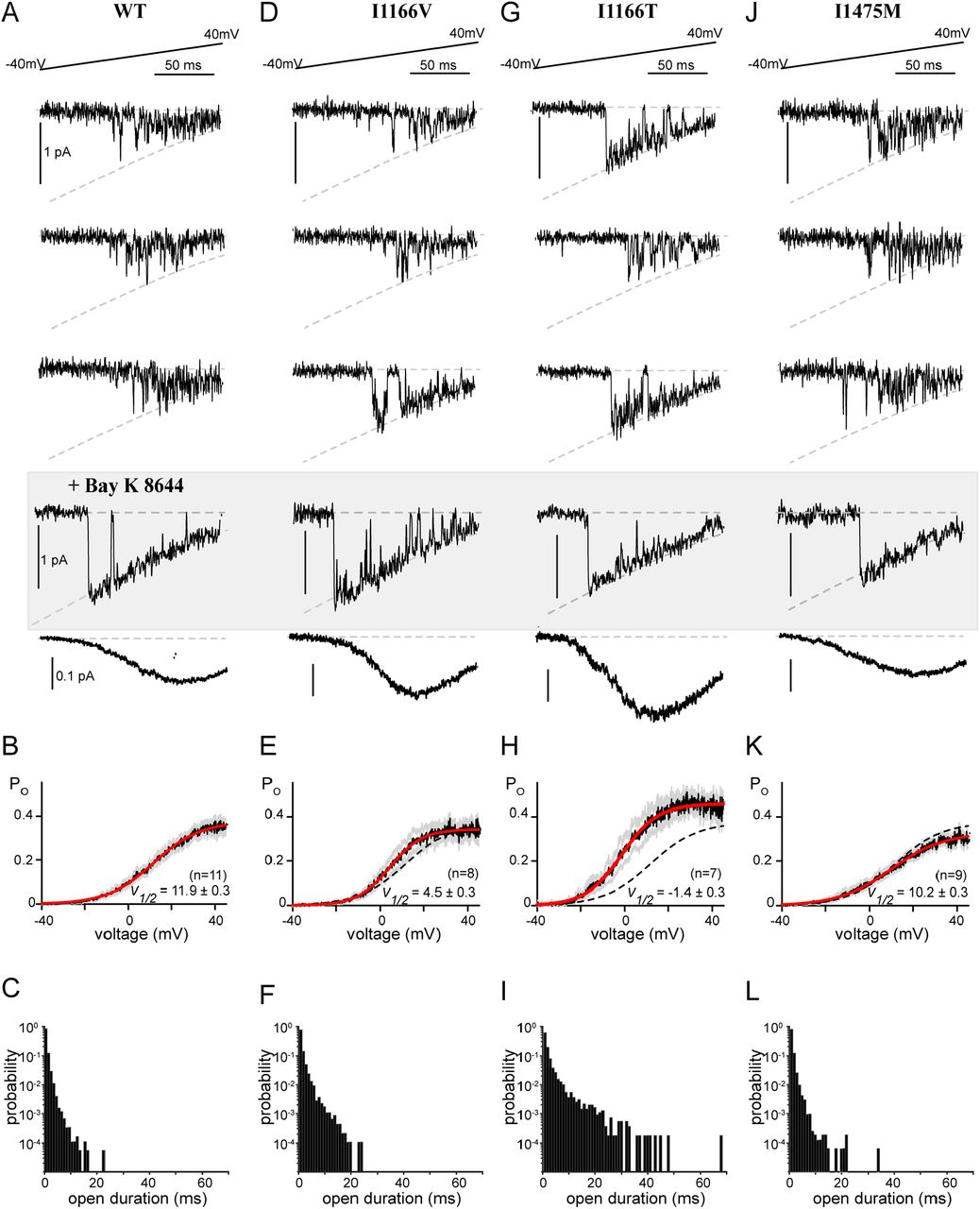
Single channel recordings reveal altered gating
(A) Top: Exemplar single channel traces in response to a voltage ramp. The ramp was applied from a voltage of -80 mV to 70 mV, however data is displayed from -40 mV to 40 mV for visualization of the single channel properties. Channel openings are seen as the downward deflections, with the dashed line indicating the single channel current as a function of voltage, as defined by the GHK equation. Middle shaded box: Bay K8644 was added at the conclusion of the experiment to make it easier to count the number of channels in the patch. Bottom: Averaging numerous sweeps generates the average current-voltage relationship for each cell. (B) Quantification of the open probability as a function of voltage. Black indicates the average open probability from multiple cells, gray displays the SEM and red displays the Boltzmann fit to the final data. V1/2 is displayed as ± the 95% confidence interval of the fit as determined by Prism (GraphPad). (C) Open duration histograms demonstrating the probability of opening events lasting for the indicated duration. (D) Exemplar single channel traces for the I1166V mutation demonstrate no change in channel conductance (dashed curve). However, while the majority of traces exhibited mode 1 gating (top 2 traces), cells would periodically display mode 2 gating (compare bottom exemplar with the Bay K 8644 exemplars). (E) Consistent with brief sojourns into mode 2; the average open probability as a function of voltage was somewhat left shifted as compared to WT (dashed line). Format as in B. (F) The brief periods of mode 2 gating can be seen as an increased probability of longer open durations. (G) Exemplar traces demonstrate significant mode 2 behavior for channels containing I1166T. (H) The enhanced mode 2 gating results in a large left shift in the open probability curve (red) as compared to WT (dashed line), and an overall increase in PO across voltages. Format as in B. (I) Open duration histograms demonstrate a significant proportion of mode 2 openings, with durations extending beyond 40 ms. (J-L) No change in single channel parameters were identified in channels harboring I1475M.
We next applied the ramp protocol to channels harboring the I1166V mutation, which produced a modest left shift in channel activation in whole cell experiments (Fig. 1). Indeed, single channel measurements recapitulated the left shift in activation (Fig. 4D, E), without significant overall changes to conductance or the maximal PO (PO,max). Interestingly, the left shift in activation was primarily due to periodic entry into mode 2. While WT CaV1.2 channels will occasionally enter mode 2 without perturbation, generally application of an antagonist such as BayK 8644 (Fig. 4, gray box) is required to cause mode 2 gating. The mode 2 gating caused by I1166V is demonstrated in the exemplar traces displayed (Fig. 4D), where sweeps from the same recording switch from a mode 1 characteristic (top 2 traces) to mode 2 (bottom trace). This is further quantified by the open duration histogram (Fig. 4F), which displays an enhanced number of long duration openings. This mode 2 switching was even more significant when we looked at the I1166T mutation (Fig. 4G). There was again no change in channel conductance, however channels harboring the I1166T mutation spent a significant amount of time in mode 2, resulting in a large left shift in channel activation as well as an overall increase in PO,max (Fig. 4H). Open duration histograms demonstrate a considerable shift to long duration openings, with durations extending beyond 40 ms (Fig. 4I). This remarkable behavior results in a substantial increase in channel opening across multiple voltages and correlates with the severity of the patient phenotype. Interestingly, while this increased PO explains the reduction in CDImax (Fig. 3D), it also points to an increase in FCDIat lower voltages as additional Ca2+ would now be available to drive channels into mode Ca. Thus, the effects of each mutation on these two parameters are unlikely to be entirely independent.
Finally, we turned our attention to the I1475M mutation, which displayed a loss of CDI through a deficit in CDImax despite no change in channel activation. Surprisingly, this mutation produced minimal changes to the single channel properties, with no significant change in conductance, V1/2 of activation or open duration (Fig. 4 J-L). While we did note a slight decrease in PO,max (Fig. 4K), this small effect cannot account for the CDI changes produced by this mutation. It therefore appears that I1475M produces a loss of CDI through a novel mechanism.
A closer look at the neuronal effects of CaV1.2 mutations
The majority of CaV1.2 mutations reported in patients, and all in this study, are associated with cardiac symptoms (27, 38). However, only a select few are associated with the severe neurological phenotypes identified among TS patients (27, 38). While alterations in CDI, VDI and activation represent a recognized mechanism for inducing LQTS (19, 31), the same cannot be said for the developmental delay or ASD seen in some CaV1.2 channelopathy patients. Of the mutations evaluated in this study, only I1166T has been described as causing a neurological deficit in patients. Interestingly, this mutation also results in a dramatic increase in channel activation, both through a left shift in the voltage dependance of activation, and enhanced entry into mode 2 gating (Fig. 4H). We therefore wanted to probe whether these biophysical properties were sufficient to alter the response of the channels to a neuronal action potential. By expressing the channels within HEK 293 cells and applying a neuronal electrical stimulus, we observe only those effects due directly to channel gating, eliminating the contribution of downstream signaling, channel trafficking or developmental changes. In order to more closely match physiological conditions, experiments were carried out in an external solution containing 1.8 mM Ca2+, and an internal solution containing 1 mM EGTA which permits more physiological intracellular Ca2+ buffering (39). In addition, we utilized the β1B subunit such that both CDI and VDI are enabled (32). Ca2+ currents were then recorded in response to a 40 Hz train of neuronal action potentials (APs), and the amplitude of each Ca2+ response was measured (Fig. 5A). For WT channels, the peak current in response to the repetitive APs decayed slightly over the course of the stimulus (Fig. 5B). However, when CaV1.2 channels harboring the I1166T mutation were exposed to the same stimulus, the amplitude decay (as normalized to the first peak) was significantly increased (Fig. 5C), demonstrating a clear difference as compared to WT channels. The same protocol applied to I1166V, I1475M, L762F or E1496K resulted in amplitudes identical to WT channels (Fig. 5D-G). Thus, unlike mutations which selectively alter CDI or VDI, the large changes in channel activation of I1166T are sufficient to produce a differential response to a neuronal signal.
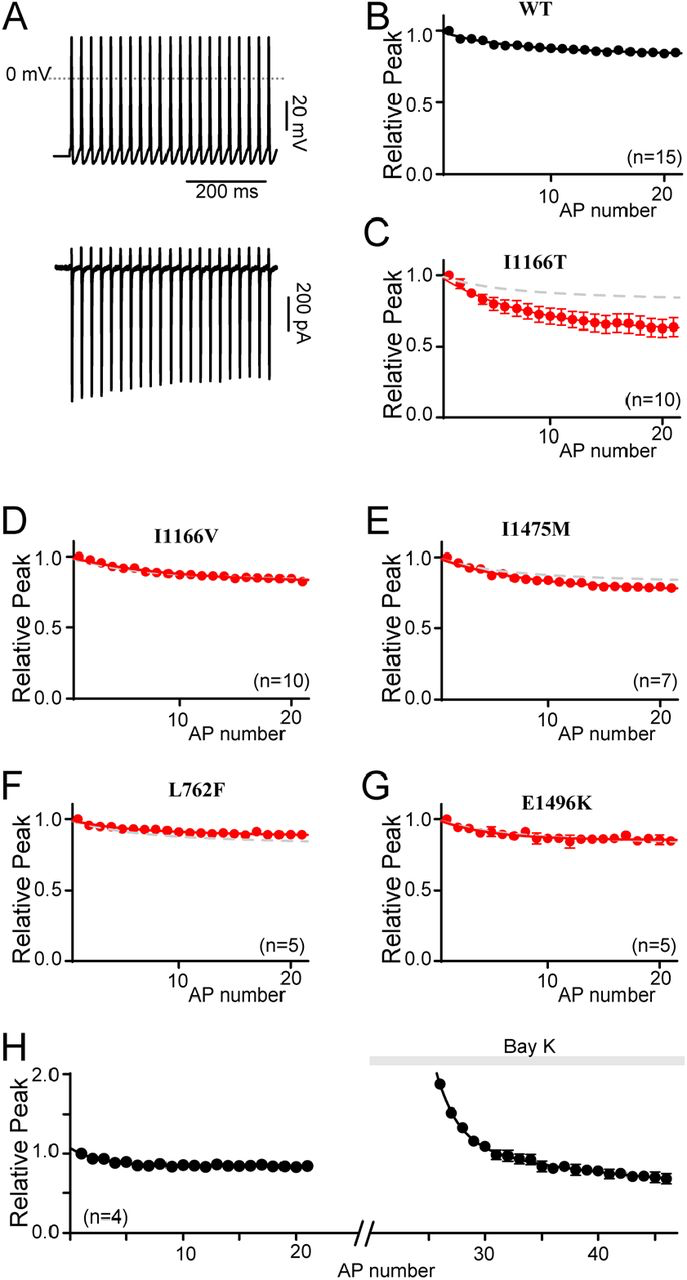
Differential responses to a neuronal stimulus
(A) Exemplar Ca2+ current response (bottom) to a 40 Hz train of neuronal action potentials (top).
(B) Normalized peak current amplitude plotted as a function of AP number within the train reveal a modest decay in current amplitude over time. Data is displayed ± SEM here and throughout.
(C) Normalized peak current amplitudes in channels containing I1166T (red) significantly deviate from WT (gray dashed line). (D-G) I1166V, I1475M, L762F and E1496K do not alter the normalized peak current amplitudes as compared to WT (gray dashed curve). (H) Normalized peak current amplitudes measured before and after the application of Bay K 8644 demonstrate the relative increase in current amplitude in the presence of Bay K. All currents are normalized to the first peak prior to the addition of Bay K. Data is plotted ± SEM.
While these results indicate a unique feature for the I1166T mutation, they also appear to violate the prior results which showed a loss of CDI for channels harboring the I1166T mutation. Here, I1166T appears to enhance inactivation of the current in response to a train of APs. However, under the conditions of this experiment it is unlikely that maximal CDI is achieved during the short duration of the neuronal APs, even after repetitive stimulation. Instead, we postulated that the I1166T mutation caused a large increase in the amplitude of the initial peak, however the need to normalize the data in order to compare across cells removed this component. The subsequent enhanced decay of the current through channels harboring I1166T would then result from an augmented Ca2+ driving force (increased FCDI) due to the mode 2 openings of the channel. Thus, the effects of I1166T are due to a complex interplay between a deficit in CDIMax (Fig. 3D) and an increase in FCDI. To demonstrate this, we considered that the effect of the I1166T mutation is very similar to the effect of BayK 8644 when applied to WT channels. This allowed us to directly examine the impact of enhancing entry into mode 2 within the same cell. Indeed, when the AP train was applied to WT cells before and after application of Bay K 8644, a clear increase in current amplitude due to Bay K 8644 application can be seen (Fig. 5H). The enhanced decay of the current then brings the amplitude back down towards the baseline. Thus, the cumulative effect of the I1166T mutation on CaV1.2 current amplitude is that of a gain-of-function, as predicted by the increased activation of the mutant channel.
Discussion
Mutations in CaV1.2 have been linked to severe cardiac and neurological disorders which are often fatal in early childhood. The first two mutations described, G406R and G402S produced profound phenotypes in patients despite the fact that they occurred within a mutually exclusive exon resulting in expression in only a fraction of CaV1.2 channel variants (7, 8). This severe phenotype at low expression levels may be partly due to the fact that the mutations cause significant changes to channel activation, VDI and CDI (22, 40). The mutations evaluated in the current study; however, are each expressed in a constitutive exon, resulting in much higher expression in the patient. Thus, the more selective effects on channel regulation are consistent with the patient phenotypes; severe mutations such as G406R would likely be embryonic lethal at these higher expression levels(22). Nonetheless, the gating effect of the I1166T mutation is quite severe, even in the absence of a VDI deficit. This observation fits with the relative severity of this mutation in patients, where I1166T produced the most severe phenotype of those in this study (13, 14, 16). It is also the only mutation in this study which correlates with the severe multisystem disorder described in the original TS patients harboring the G406R mutation.
Thus far, attempts to correlate biophysical changes due to CaV1.2 mutations and patient phenotype have not yielded definitive results (14). This may be partly due to the comparison of experimental results across multiple conditions from different labs, or from the limited number of in-depth mechanistic studies which distinguish between different forms of channel regulation. It has also been suggested that neuronal effects of some CaV1.2 mutations may be independent of Ca2+ permeation (41). We therefore sought to elucidate the detailed changes in channel gating due to CaV1.2 mutations. We evaluated several mutations at both the whole-cell level (with conditions tuned for robust measurements of VDI vs. CDI), and further examined the mechanism underlying the CDI-altering mutations through quantitative Ca2+ imaging and single channel recordings. Within these parameters, we do indeed find that only the multisystem I1166T mutation produces a large increase in channel activation. Moreover, our results indicate that the gating changes of this mutation are sufficient to reproduce a change in neurological response even in the absence of downstream signaling or neuron-specific regulation (Fig. 5). Interestingly, both G406R and I1166T share these features of enhanced channel activation and neurological phenotypes. On the other hand, CDI and VDI deficits are present in multiple cardiac-selective mutations which did not produce an overt change in current response to a neuronal stimulus. We therefore postulate that mutations which decrease CDI or VDI, or enhance channel activation are causative of LQTS, however, only those with significant enhancement of channel activation are likely to result in ASD or neurodevelopmental disorders (Fig. 6). Interestingly, this result is consistent with mistuning of a voltage-dependent conformational change which has been previously identified as important for the pathogenesis of the canonical TS mutations (42); thus, in a neuron the full impact of the TS mutations may proceed from more than one pathogenic mechanism, with enhanced activation as a common feature (27).
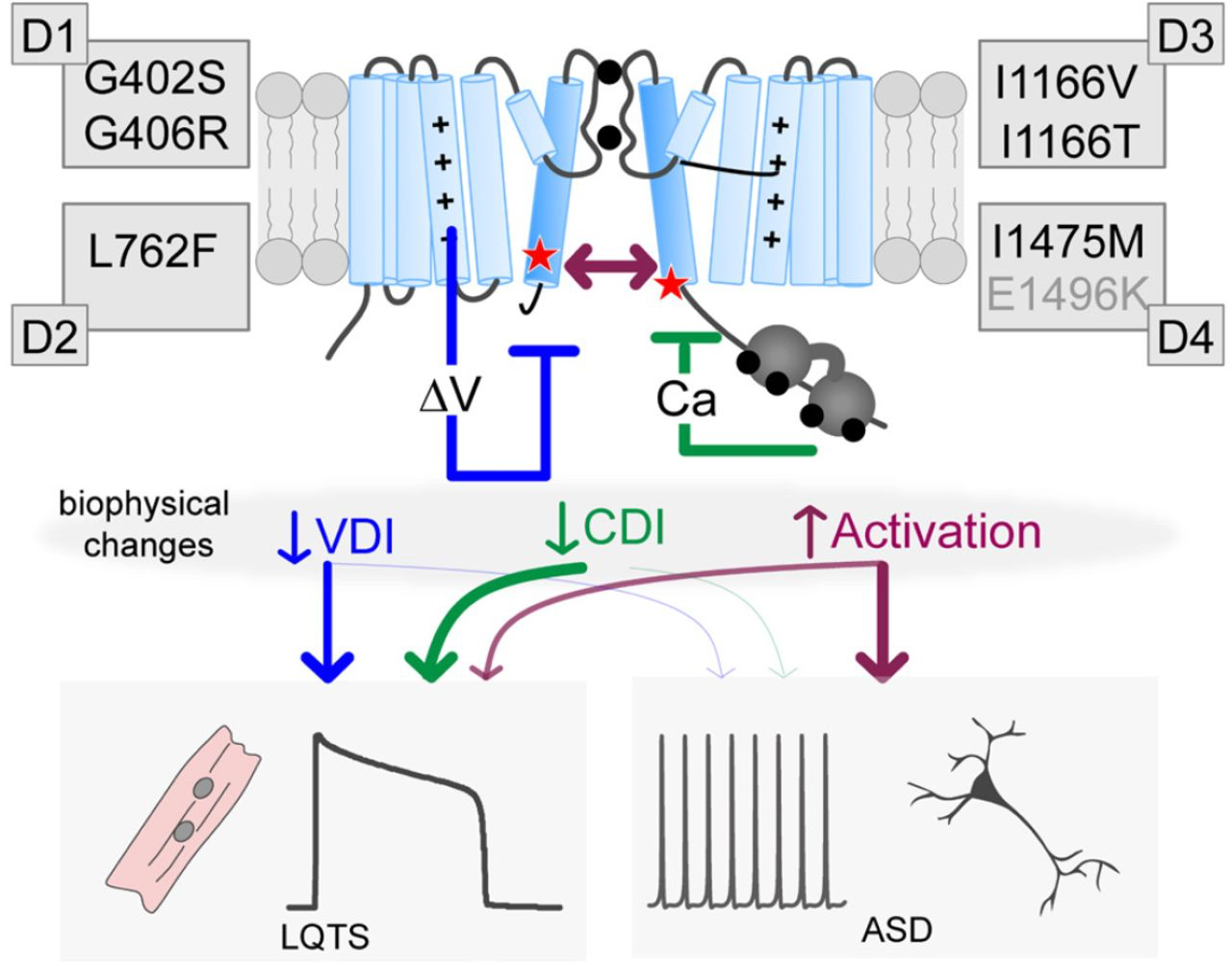
Distinct biophysical changes yield divergent pathological outcomes
Cartoon depicting the potential impact of the mutations. Each mutation can impact VDI (blue), CDI (green) and/or activation (purple), leading to LQTS. However, only large increases in channel activation appear to contribute to neurodevelopmental deficits. Note the E1496K mutation is labeled in gray as it resides outside the S6 region, nonetheless, it fits within this scheme as a VDI altering mutation.
In addition to providing insight into the mechanism underlying the pathogenesis of CaV1.2 mutations, this study also expands our knowledge of the role of the S6 region in channel gating. While pathogenic mutations in CaV1.2 have been identified across multiple distinct channel regions, many appear to cluster near a distal S6 and exert their impact through a change in channel gating. Interestingly, this S6 pattern is emerging as a common theme across multiple ion channels (7, 43–46), making S6-opathies an important and growing class of ion channel diseases (43). All but one of the mutations in this study occurs within a distal S6 region, and our results demonstrate that these S6 mutations can exert independent and separable effects on VDI, CDI and channel activation. The separable effects on VDI are consistent with previous models of VDI which include the distal S6 region as a locus for binding of a ‘hinged-lid’ (23). More surprising; however, is the selective effect of I1475M on CDI. Prior work has suggested that the impact of S6 mutations on CDI is often secondary to a change in the activation energy required to open the channel (23, 47). Here, we demonstrate that this is not always the case, and the S6 may participate in transducing CDI more directly. Interestingly, this effect was only seen in the distal IVS6 region, which connects the transmembrane region to the C-tail of the channel, where the Ca2+ sensor CaM resides on the channel. As such, it may be that the I1475M mutation disrupts the transduction of CDI following Ca2+ binding to CaM.
While the lack of changes in channel activation is remarkable for I1475M, the opposite is true for I1166T. For this mutation, a dramatic increase in mode 2 gating was apparent in the single channel recordings, resulting in a significant left shift in channel activation, and increased open probability across voltages (Fig. 4G-I). Previous studies have also suggested increased mode 2 gating in the context of a TS mutation. Erxleben et. al. showed an increase in mode 2 gating in CaV1.2 channels harboring the G406R mutation; however, in this case the entry into mode 2 was thought to be controlled by an aberrant CaMKII phosphorylation site (48). However, insertion of the I1166T mutation does not form a consensus CaMKII sequence, and insertion of a V at the same site also increased the propensity for mode 2 gating, making it unlikely this mutation represents a phosphorylopathy. However, it is interesting to note that I1166 resides within a ring of hydrophobic S6 residues which are believed to stabilize the closed state of the channel (24). Mutation of the hydrophobic ‘I’ to a neutral ‘T’ residue may play a role in the enhanced opening of channels harboring the I1166T mutation. The far lesser effect of the I1166V mutation may then reflect the lack of a significant change in hydrophobicity, but nonetheless points to the importance of the I1166 residue in stabilizing the closed configuration as this mutation also produced a left shift and enhanced mode 2 gating, albeit to a far lesser extent. Likewise, the lack of an activation shift for the I1475M mutation, which resides within the IVS6 hydrophobic region, may fit with the preservation of the hydrophobicity of this residue.
It should be noted that whole cell recordings have previously been carried out on the mutations described in this study (13, 14, 16), sometimes with results which are inconsistent with the current study. In particular, E1496K and I1475M were previously reported to produce a left shift in channel activation (13). While it is not clear why these results differ, experimental conditions were quite different in that prior experiments were carried out in oocytes and activation curves were determined by a fit to the current-voltage relation. Given the important implications of the lack of activation shift for I1475M, our results from tail current protocols were confirmed by single channel recordings, demonstrating no activation shift under our experimental conditions.
Overall, this study provides an in-depth analysis of the biophysical mechanisms underlying select CaV1.2 channelopathic mutations. By focusing primarily on mutations within the S6 region, this study furnishes an in depth understanding of the role of this channel segment in both activation and inactivation. Given the growing number of S6-opathy mutations being identified, such structure-function understanding may provide insight into a variety of channelopathies.