
Amyotrophic lateral sclerosis: a neurodegenerative disorder poised for successful therapeutic translation

By Richard J. Mead, Ning Shan, H. Joseph Reiser, Fiona Marshall, and Pamela J. Shaw
Excerpt from the article published in Nature Reviews Drug Discovery, 21 December 2022. DOI: https://doi.org/10.1038/s41573-022-00612-2
Abstract
Amyotrophic lateral sclerosis (ALS) is a devastating disease caused by degeneration of motor neurons. As with all major neurodegenerative disorders, development of disease-modifying therapies has proven challenging for multiple reasons. Nevertheless, ALS is one of the few neurodegenerative diseases for which disease-modifying therapies are approved. Significant discoveries and advances have been made in ALS preclinical models, genetics, pathology, biomarkers, imaging and clinical readouts over the last 10–15 years. At the same time, novel therapeutic paradigms are being applied in areas of high unmet medical need, including neurodegenerative disorders. These developments have evolved our knowledge base, allowing identification of targeted candidate therapies for ALS with diverse mechanisms of action. In this Review, we discuss how this advanced knowledge, aligned with new approaches, can enable effective translation of therapeutic agents from preclinical studies through to clinical benefit for patients with ALS. We anticipate that this approach in ALS will also positively impact the field of drug discovery for neurodegenerative disorders more broadly.
Key points
- Amyotrophic lateral sclerosis (ALS), with a lifetime risk of ~1/350, represents an area of huge unmet need and is a useful model of neurodegeneration, with measurable changes in motor function over a relatively short time frame.
- The field of ALS has advanced significantly over the last decade, with rapid progress in understanding the genetic architecture and the pathophysiological mechanisms of the disease, and in the development of robust, exploitable preclinical model systems.
- Potential biomarkers of phenotypic conversion, target engagement and therapeutic efficacy have now emerged. Plasma and cerebrospinal fluid (CSF) neurofilament protein levels look particularly promising and may improve the efficiency of future clinical trials and allow identification of responder subgroups.
- The identification of several biological pathways with the potential to be tackled therapeutically has generated a promising pipeline of preclinical approaches and clinical trials.
- Genetic therapy trials are now poised for successful translation. In addition, combination therapies or therapies with the potential to ameliorate several pathophysiological mechanisms contributing to motor neuron injury are now being evaluated.
- Recent innovations in trial design are poised to enhance outcome measures, and patient selection and randomization, while minimizing the impact of disease heterogeneity and increasing statistical power.
Introduction
Amyotrophic lateral sclerosis (ALS), also known as motor neuron disease, is a devastating neurodegenerative disorder in which degeneration of the upper motor neurons in the motor cortex and lower motor neurons in the brainstem and spinal cord cause progressive denervation of voluntary muscles. ALS occurs globally, with an incidence of approximately 2 per 100,000 person-years, a prevalence of 6–9 per 100,000 persons (refs. 1,2) and a lifetime risk of approximately 1 in 350 (ref. 3). There is evidence that the condition is increasing in incidence1,4. This may be partially accounted for by changing population demographics, and improved clinical services supporting accurate diagnosis. A family history of ALS is present in 5–10% of affected individuals, usually with an autosomal dominant inheritance pattern. However, systematic genetic testing has revealed the presence of an identifiable genetic cause in a larger proportion of patients with ALS (see below). Advancing failure of the neuromuscular system causes progressive weakness of the muscles in the upper and lower limbs, as well as the bulbar and respiratory muscles. The rate of disease progression is variable, but the majority of patients die from neuromuscular respiratory failure within 2–3 years of symptom onset5. There is an overlap between ALS and frontotemporal dementia (FTD) — approximately 5% of patients with ALS develop overt features of FTD, but detailed neuropsychological evaluation reveals more subtle disturbance of frontal and temporal lobe function in up to 50% of patients6,7.
Pathologically the key features of ALS include loss of upper and lower motor neuron cell bodies and degeneration of the corticobulbar/corticospinal tracts and lower motor neuron axons, with denervation changes within muscles. In most patients (~97%), ALS is a TAR DNA-binding protein 43 (TDP-43) proteinopathy. As motor neurons become injured there is loss of the TDP-43 protein from the nucleus, with cytoplasmic aggregation into structures with compact or skein-like morphology8,9,10. However, there is also pathological heterogeneity: ALS caused by mutations in the SOD1 (Cu–Zn superoxide dismutase) and FUS (fused in sarcoma) genes is not a TDP-43 proteinopathy, although cytoplasmic protein aggregates of different composition are present11,12,13,14. In addition, the most common genetic subtype of ALS, caused by intronic hexanucleotide GGGGCC expansions in the C9orf72 gene, does have TDP-43 mislocalization, but has additional p62-positive protein aggregates caused by pathological dipeptide repeat proteins (DPRs)15.
The diagnosis of ALS is made by exclusion of mimic disorders16. Diagnostic investigations usually include a battery of blood tests, imaging of the brain and spine to exclude structural pathology and a neurophysiological assessment. Diagnostic criteria include the revised El Escorial criteria17, the Awaji Shima criteria18 and most recently the simplified Gold Coast criteria19.
Supplementary Table 1 highlights currently available symptomatic and disease-modifying therapies available for ALS. More than 60 compounds, with different mechanisms of action, have been evaluated in clinical trials in ALS20, but only three of these have been approved for clinical use: riluzole, edaravone and AMX0035. Riluzole was the first FDA-approved therapy for ALS21,22,23. It is considered to reduce glutamate release into the synaptic cleft by blocking voltage-gated sodium channels on presynaptic neurons, and thus to ameliorate excitotoxicity24,25. The disease-modifying effect has been regarded as modest, the initial trial results indicating a prolongation of survival by approximately 3 months on average. However, population studies comparing patients who did and did not receive riluzole treatment documented substantially larger increases in survival, ranging from 6 to 19 months26.
Edaravone, an antioxidant agent administered intravenously for 14 days per month, was evaluated in several trials in Japan27,28. Over a 6-month trial period there was evidence that edaravone slowed disability progression in selected patients early after disease onset and with rapid disease progression27. It is not yet known whether there is an effect on survival. Edaravone was approved for the treatment of ALS in Japan in 2015, and has been approved by the FDA (2017) as well as regulatory authorities in Canada (2018) and other Asian countries including China. To date, edaravone has not been approved by the EMA. An oral formulation of edaravone has recently been approved in the USA and will probably rapidly replace the intravenous formulation in the treatment of patients with ALS. The FDA recently approved AMX0035, developed by Amylyx, for the treatment of ALS after convening an expert panel on two occasions to consider the complex phase II trial data. AMX0035 is a fixed-dose combination of taurursodiol and sodium phenylbutyrate that is considered to mitigate mitochondrial dysfunction and endoplasmic reticulum (ER) stress.
Symptomatic treatment of ALS includes both pharmacological and non-pharmacological interventions as shown in Supplementary Table 1. Most patients with ALS die from neuromuscular respiratory failure. Intervening with non-invasive ventilation when significant weakness of the respiratory muscles develops has a significant impact in improving both life expectancy and quality of life29,30.
In this Review, we describe advances in the molecular subclassification of ALS based on genetics. We review new insights into the pathophysiology of ALS which show promise for drug discovery. We describe advances in preclinical disease modelling which improve the prospects for identification of effective neuroprotective therapies. We describe the current landscape of clinical trials for ALS and summarize the preclinical pipeline of potential therapeutic agents. Finally, we discuss reasons for the relatively high failure rate of ALS clinical trials to date, and suggest strategies to address the huge unmet need for new ALS therapies.
ALS pathophysiology
Motor neuron injury in ALS is considered to result from multiple interacting pathophysiological mechanisms that culminate in widespread network disruption. The heterogeneity of ALS suggests that different mechanisms may play more or less prominent roles in individual patients. Advances in preclinical disease modelling (Box 1) are needed to capture this complexity. To develop neuroprotective therapies, an advantageous approach could include upstream targeting of the initiating factor (for example, by gene silencing or gene replacement), and one or more drugs that ameliorate multiple facets of the disease pathophysiology. This section reviews recently emerging insights into contributory pathophysiological mechanisms (Fig. 1) and highlights novel therapeutic strategies.
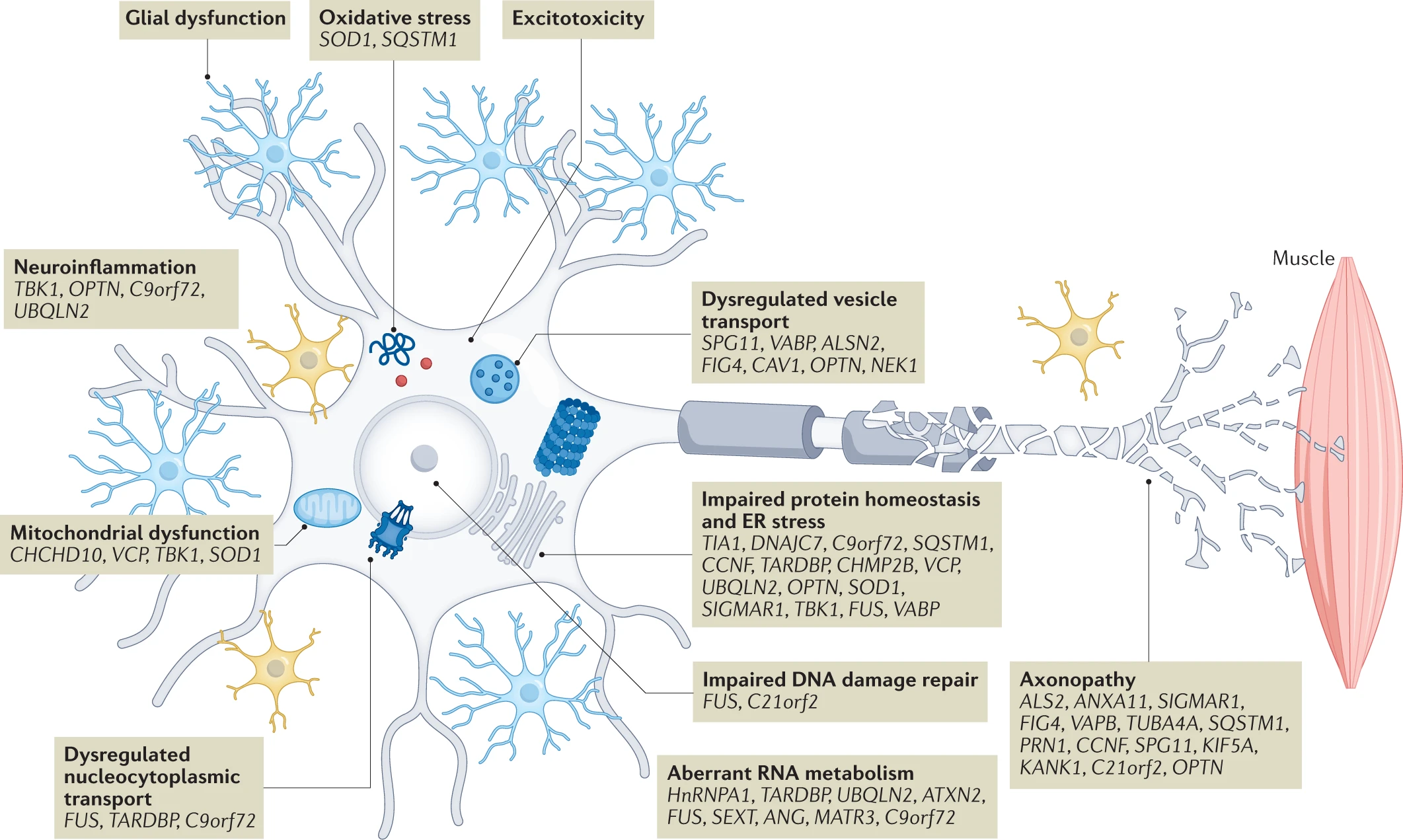
ALS pathophysiology, genetic causes and risk factors.
Advances in large-scale genomic analysis have uncovered a variety of causative genes and risk factors for amyotrophic lateral sclerosis (ALS). These gene variants map onto key pathogenic mechanisms relevant to all motor neuron cellular compartments as well as neighbouring cells such as glia and interneurons. In this way, these mechanisms are genetically validated, enabling a greater confidence in their targeting for therapeutic benefit. Some of these mechanisms have emerged only in recent years due to new genetic information, including gene changes highlighting dysregulation of RNA processing and metabolism. There is significant overlap of some genes with those found in closely related disorders such as frontotemporal dementia (for example, C9orf72, CHCHD10, SQSTM1, TBK1, CCNF, FUS, TARDBP, OPTN, UBQLN2, TUBA4A, ATAXN2, VCP and CHMP2B). This suggests a closer relationship to broader neurodegenerative disorders, and indeed many of the pathways depicted are relevant in, for example, Alzheimer disease. ER, endoplasmic reticulum. For a complete list of the ALS loci, genes and associated proteins, see Supplementary Table 2.
Box 1 Advances in preclinical disease modelling in ALS Rational preclinical screening cascades for drug discovery rely on disease models which enable some level of confidence in translation of preclinical efficacy to the clinic. For neurodegeneration in particular, the identification of models which predict translational success is still a significant issue, with >90% of trials in neurodegenerative diseases failing for lack of efficacy240. Amyotrophic lateral sclerosis (ALS) is an interesting case as, until recently, it was the only neurodegenerative disease for which disease-modifying therapies were available to validate disease models. Recent advances, such as the ability to create patient-derived central nervous system (CNS) cell types in the laboratory and the advent of a battery of new genetic models of ALS, hold great promise for improving efficacy predictions. Historical approach The identification of mutations in SOD1 in 1993 (refs. 31,241) enabled the generation of the first SOD1-mutant transgenic mouse models of ALS242. These models show a predominant motor phenotype with spinal motor neuron degeneration and loss, despite ubiquitous high-level over-expression of mutant SOD1 via its endogenous promoter243. Guidelines on the use of ALS models, particularly the SOD1-mutant model, were published in 2010 (ref. 244). Testing of therapeutic agents previously shown to extend survival using these new approaches failed to show any benefit243 suggesting that much of the published literature was not reproducible. Further limitations relate to the fact that this model is based on a genetic mutation which does not broadly represent ALS cases, best evidenced by a lack of TDP-43 proteinopathy in SOD1-mutant mice245, in contrast to the vast majority (>97%) of patients with ALS12. However, the SOD1-mutant rodent models still have value as part of a rational pharmacology screening cascade. They display motor neuron degeneration driving a disease phenotype of muscle weakness and atrophy, as in clinical ALS. They recapitulate, and indeed have been used to validate, many of the disease mechanisms implicated in human disease (Fig. 1) and in some cases have been shown to respond to approved therapies such as riluzole and edaravone246,247. In this way, the model is best viewed as a test bed for therapeutic approaches that target specific disease mechanisms hypothesized to be driving the disease phenotype. This allows the generation of pharmacokinetic–pharmacodynamic data in relation to mechanism as well as the development of both translational (for example, compound muscle action potential, plasma neurofilament light) and target engagement biomarkers which can be used to enhance translational approaches. New genetic mouse models with broader relevance The explosion in genetic characterization of ALS (Table 1; Supplementary Table 2) has enabled new models to be developed. With respect to mouse models, the most significant work relates to mutations in TDP-43, FUS and C9orf72. TDP-43 proteinopathy is a pathological hallmark in the majority of patients with ALS, including those with sporadic ALS8. Subsequently, mutations in TARDBP were found in a small proportion of patients with ALS248. Multiple transgenic models were generated248 and it quickly became apparent that, unlike SOD1, over-expression of either mutant or wild-type TDP-43 caused neurodegeneration and, where that over-expression surpassed a particular threshold, neurodegeneration was particularly rapid249. In addition, evidence accumulated that loss of TDP-43 could cause neurodegeneration250. Models designed to approximate physiological expression of Q331K-mutant TDP-43 showed a milder phenotype without attendant TDP-43 pathology251,252, suggesting that overt TDP-43 pathology may be a later stage event. A knock-in model with the same mutation recapitulated some of the features of the mild over-expression models, including a lack of TDP-43 proteinopathy and a counter-intuitive increase in nuclear TDP-43 (ref. 253). Further MRI-guided investigation has implicated interneuron loss and highlighted similarities to human ALS254. Of note, a humanized FUS model introduced an exon-skipping mutation found in patients with ALS into the genome of mice, resulting in a frameshift mutation which was expressed at physiological levels255. Interestingly, this model also demonstrated neurodegeneration without typical FUS-related proteinopathy, but did recapitulate FUS mislocalization from nucleus to cytoplasm. These data, combined with those in the more physiological TDP-43 models, suggest that typical ALS inclusion proteinopathy may be a late-stage event which should therefore be targeted therapeutically with caution. Although these humanized models are welcome and provide useful mechanistic insights, their use as preclinical models for evaluation of potential neuroprotective agents may still be limited due to the mild phenotypes. The use of low expressing transgenic TDP-43 mice which phenocopy the knock-in TDP-43 models but in a shorter timescale may be a better approach252. Given the prevalence of C9orf72 repeat expansions in human ALS, there is great interest in creating mice with this genetic lesion. Multiple C9orf72-transgenic mice strains have been developed which carry a BAC construct derived from a patient with C9orf72-mutant ALS. Many of these models show a molecular phenotype, consisting of RNA foci and dipeptide repeat proteins and in general either no or limited motor and cognitive phenotypes256,257. One model on the FVB background was reported to have a profound combined motor and cognitive phenotype258, which has been replicated in some laboratories259, but not others260. Emergence of patient-derived cellular models The ability to isolate, culture and re-programme patient-derived cells such as fibroblasts into CNS cells has added a new dimension to many fields of drug development including ALS261. Two main routes exist for such an approach. The use of induced pluripotent stem cells (iPSCs) has been explored in multiple laboratories with a focus on generation of patient-derived motor neurons from patients with mutations in TARDBP, FUS, SOD1 and C9orf72 or sporadic disease. To date, these have been used in repurposing screens of relatively small libraries, targeting, for example, TDP-43 proteinopathy, protein dyshomeostasis and hyperexcitability261. Some of the molecules identified using these repurposing approaches have entered clinical trials, including bosutinib262 and ropinirole263. However, the bosutinib trial was terminated in 2022. The focus on monocultured motor neurons as a screening approach limits the contribution of non-neuronal cells to the observed phenotypes. It is well described in ALS that neighbouring cells, such as astrocytes and microglia, have a profound effect on neurodegenerative phenotypes and neuronal survival264. In addition, the use of iPSC technology generally leads to a loss of the cellular ageing signature265. A second approach involves the trans-differentiation of patient fibroblasts to neural progenitors which can then be further differentiated to neurons or astrocytes124. These astrocytes show the predicted toxicity towards cocultured motor neurons and appear to maintain the associated ageing phenotype219. The next stage in incorporating the physiological relevance and complexity in such models is to introduce 3D organoid culture. Recent efforts have, for example, led to the formation of patient-derived organoids with impaired neuromuscular junctions266. However, further work is needed to validate and obtain regulatory approval for the use of these human cellular models as tools for effective ALS drug discovery.
Genetic architecture
Since the discovery of SOD1 mutations as a cause of familial ALS (fALS) in 1993 (ref. 31), our understanding of the genetic architecture of ALS has increased substantially. ALS is an archetypal complex disease which has a monogenic cause in ~10–21% of patients, but in the majority of affected individuals is determined by an interaction of multiple genetic and environmental risk factors.
Of patients with ALS, 5–10% have familial disease, usually with a Mendelian autosomal dominant pattern of inheritance. The genetic cause in 60–70% of individuals with fALS has now been identified32. In 90–95% of patients with ALS, the disease is currently classified as sporadic ALS (sALS), but genetic factors are considered important even in the absence of a family history. The heritability of sALS has been estimated at approximately 50%3,33. Analyses of large genome-wide association studies (GWAS) of patients apparently with sALS have indicated that the genetic architecture of ALS is based predominantly on rare variants34. Thus, the traditional subclassification of ALS into fALS and sALS is now recognized as overly simplistic and the field is moving towards a more accurate molecular subclassification based on the identification of risk genes.
More than 30 genes have been identified (Table 1; Supplementary Table 2) which are causative in or confer an increased risk of the development of ALS. Four genes account for the disease in up to 70% of patients with fALS, at least in European populations: C9orf72, SOD1, TARDBPand FUS. A recent extensive GWAS involving a meta-analysis of 29,612 patients with ALS and 122,656 controls identified 15 new genetic risk loci33. Interestingly, the risk of developing ALS and the factors controlling severity of disease (age at onset and speed of progression) appear to be genetically independent. The identified risk genes converge on several key biological pathways including: oxidative stress; dysregulation of mitochondrial function, protein homeostasis, RNA processing, axonal transport and nucleocytoplasmic transport (NCT); neuroinflammation; excitotoxicity; and DNA damage (Fig. 1). These pathways should be intensively interrogated for new therapeutic targets.
| ALS locus numbera | Gene | Encoded protein | Chromosomal location | Inheritance | Phenotypic features | Protein function: disease mechanisms |
|---|---|---|---|---|---|---|
| ALS1 | SOD1 | Cu-Zn superoxide dismutase | 21q22.11 | AD (AR) | Adult-onset, usually limb onset; not associated with dementia; not a TDP-43 proteinopathy | Dismutates superoxide free radicals: oxidative stress; protein aggregation; mitochondrial dysfunction; axonal transport defects; proteasome impairment; glial dysfunction |
| ALS2 | ALS2 | Alsin | 2q33.1 | AR | Infantile and juvenile-onset, slowly progressive ALS mainly affecting upper motor neurons | Intracellular trafficking |
| ALS4 | SETX | Senataxin | 9q34.13 | AD | Juvenile-onset, slowly progressive ALS | RNA processing |
| ALS5 | SPG11 | Spatacsin | 15q21.1 | AR | Juvenile-onset, slowly progressive ALS mainly affecting upper motor neurons | Vesicle trafficking; axonal defects |
| ALS6 | FUS | Fused in sarcoma RNA binding protein (component of the hnRNP complex) | 16p11.2 | AD (AR) | Large variation in the age at disease onset, but with a median age younger than for sporadic ALS; typical or atypical ALS and FTD | RNA processing; DNA damage repair defects; nucleocytoplasmic transport defects; stress granule function; protein aggregation |
| ALS8 | VAPB | Vesicle-associated membrane protein | 20q13. 32 | AD | Adult-onset, typical or atypical ALS | Proteasome impairment; intracellular trafficking |
| ALS9 | ANG | Angiogenin | 14q11.2 | AD | Adult-onset, typical ALS and FTD | RNA processing |
| ALS10 | TARDBP | TDP-43 | 1p36.22 | AD | Adult-onset, typical ALS not associated with overt cognitive dysfunction; limb or bulbar onset, considerable variation in age at onset and rapidity of disease course | RNA processing; nucleocytoplasmic transport defects; stress granule function; protein aggregation |
| ALS11 | FIG4 | Polyphosphoinositide phosphatase | 6q21 | AD | Adult-onset, clinical variability with incomplete penetrance | Intracellular trafficking |
| ALS12 | OPTN | Optineurin | 10p13 | AD (AR) | Adult-onset; slowly progressive atypical amyotrophic lateral sclerosis | Autophagy; protein aggregation; inflammation; NF-κB regulation; membrane trafficking; exocytosis; vesicle transport; reorganization of actin and microtubules; cell cycle control |
| ALS13 | ATXN2 | Ataxin 2 | 12q24.12 | AD | Adult-onset, typical ALS | RNA processing |
| ALS14 | VCP | Valosin-containing protein / Transitional endoplasmic reticulum ATPase | 9p13.3 | AD/de novo | Adult-onset, typical ALS and FTD | Autophagy; proteasome impairment; defects in stress granules; protein aggregation; mitochondrial dysfunction; endoplasmic reticulum dysfunction |
| ALS15 | UBQLN2 | Ubiquilin 2 | Xp11.21 | X-linked AD | Adult or juvenile onset | Proteasome impairment; autophagy; protein aggregation; oxidative stress; axonal defects |
| ALS16 | SIGMAR1 | Sigma non-opioid intracellular receptor 1 | 9p13.3 | AD and AR | Juvenile-onset ALS associated with FTD | Proteasome impairment; intracellular trafficking |
| ALS17 | CHMP2B | Charged multivesicular body protein 2b | 3p11.2 | AD | Adult-onset, typical ALS | Autophagy; protein aggregation |
| ALS18 | PFN1 | Profilin-1 | 17p13.2k | AD | Adult-onset typical ALS | Axonal defects |
| ALS19 | ERBB4 | Receptor tyrosine-protein kinase erbB-4 | 2q34 | AD | Adult-onset, typical ALS | Neuronal development |
| ALS20 | hnRNPA1 | Heterogeneous nuclear ribonucleoprotein A1 | 12q13.13 | AD/de novo risk factor | Adult-onset typical ALS; myopathy; cognitive impairment | RNA processing |
| ALS21 | MATR3 | Matrin-3 | 5q31.2 | AD | Adult-onset; upper and lower motor neurons are affected; survival duration 2 to 12 years | RNA processing |
| ALS22 | TUBA4A | Tubulin α4A chain | 2q35 | AD | Adult-onset; frequent typical ALS presentation with some FTD-associated cases | Cytoskeleton |
| ALS23 | ANXA11 | Annexin A11 | 10q22.2 | AD | Adult-late onset, classic ALS, bulbar or limb onset | Intracellular trafficking |
| ALS24 | NEK1 | Serine–threonine protein kinase Nek1 | 4q33 | AD | Adult-onset, although clinical descriptions are scarce, typical ALS without dementia is described | Intracellular trafficking; DNA-damage response; microtubule stability |
| ALS25 | KIF5A | Kinesin heavy chain isoform 5A | 12q13.3 | AD | Adult-onset; classic ALS | Axonal defects; intracellular trafficking |
| ALS-new | GLT8D1 | Glycosyltransferase 8 domain-containing protein 1 | 3p21.1 | AD | Adult-onset, both limb onset and bulbar onset ALS in a limited number of clinically described cases | Ganglioside synthesis |
| ALS-new | TIA1 | Cytotoxic granule-associated RNA-binding protein | 2p13.3 | AD | Adult-onset; associated with both ALS and FTD | Delayed stress granule disassembly; stress granule accumulation |
| ALS-new | C21orf2 | Cilia and flagella-associated protein 410 | 21q22.3 | AD | Adult-onset typical ALS and FTD | Microtubule assembly; DNA damage response and repair; mitochondrial function; interacts with NEK1 |
| ALS-new | DNAJC7 | DnaJ heat shock protein family (Hsp40) member C7 | 17q21.2 | Unknown | Adult-onset | Protein homeostasis; protein folding and clearance of degraded proteins; protein aggregation |
| ALS-new | LGALSL | Galectin-related protein | 2p14 | Unknown | Adult-onset | Protein function is largely unknown |
| ALS-new | KANK1 | KN motif and ankyrin repeat domain-containing protein 1 | 9p24.3 | Unknown | Adult-onset | Cytoskeleton; axonopathy |
| ALS-new | CAV1 | Caveolin 1 | 7q31.2 | Unknown | Adult-onset | Intracellular and neurotrophic signalling |
| ALS-new | SPTLC1 | Serine palmitoyltransferase, long-chain base subunit 1 | 9q22.31 | AD | Juvenile-onset, variable presentation including growth retardation and cognitive dysfunction | Excess sphingolipid biosynthesis |
| ALS-new | ACSL5 | Long-chain fatty acid coenzyme A ligase 5 | 10q25.2 | Unknown | Adult-onset, rapid weight loss | Long-chain fatty acid metabolism |
| ALS-putative | ELP3 | Elongator protein 3 | 8p21 | Unknown | Adult-onset, typical ALS without dementia | Ribostasis; cytoskeletal integrity |
| ALS-putative | DCTN1 | Dynactin | 2p13 | AD | Juvenile-onset, slow progressive ALS | Axonal transport |
| ALS-putative | PARK9 | Probable cation-transporting ATPase 13A2 | 1p36.13 | AR | Juvenile-onset | Lysosome function |
| FTD-ALS1 | C9orf72 | Chromosome 9 open reading frame 72 | 9p21.2 | AD | Adult-onset, variable penetrance and clinical features | RNA processing; nucleocytoplasmic transport defects; proteasome impairment; autophagy; inflammation; protein aggregation (DPRs) |
| FTD-ALS2 | CHCHD10 | Coiled-coil–helix-coiled–coil-helix domain-containing protein 10 | 22q11.23 | AD | Adult-onset, complex phenotype including ALS, FTD, ataxia, mitochondrial myopathy, parkinsonism and sensorineural hearing loss | Mitochondrial function; synaptic dysfunction |
| FTD-ALS3 | SQSTM1 | Sequestosome-1 | 5q35.3 | AD | Adult-onset, limb onset ALS and FTD | Proteasome impairment; autophagy; protein aggregation; axonal defects; oxidative stress |
| FTD-ALS4 | TBK1 | Serine–threonine protein kinase | 12q14.2 | AD | Adult-onset; can present either as a pure motor syndrome or with cognitive/behavioural dysfunction either mild or severe enough for FTD-ALS diagnosis | Autophagy; inflammation; mitochondrial dysfunction |
| FTD-ALS5 | CCNF | Cyclin F | 16p13.3 | AD | Adult-onset | Autophagy; axonal defects; protein aggregation |
Genes identified as causative in or increasing the risk of ALS
An expanded table including references describing these genetic discoveries is shown as Supplementary Table 2. AD, autosomal dominant; ALS, amyotrophic lateral sclerosis; AR, autosomal recessive; DPR, dipeptide repeat protein; FTD, frontotemporal dementia; TDP-43, TAR DNA-binding protein 43. aALS-new represent newly described potential ALS genes that require further validation; ALS-putative represent potential genetic risk factors; FTD-ALS are genes known to be associated with both ALS and FTD.
Recent studies have shown the value of systematic genetic screening of all patients with ALS as opposed to focusing on those with familial and early-onset disease. A prospective study which undertook targeted sequencing of ALS-relevant genes showed that 21% of patients had a clinically reportable pathogenic variant and a further 21% had variants of unconfirmed significance35. Among the 13% who carried more than one variant, there was a significantly earlier age at disease onset.
Key insights emerging from recent genetic studies are highlighted in Box 2.
It is likely that genetic profiling will become standard practice and will replace the traditional classification into fALS and sALS. In this effort statistical power can be improved by reducing the search space for causative genes to areas of the genome which are functional within motor neurons36. Successful complete genetic subclassification of ALS will have a major effect on therapy development, both by targeted genetic therapy approaches and by facilitating the development of more relevant ALS models for drug screening.
Many of the genetic mutations associated with ALS are present for >50 years before disease onset. It is noteworthy that the age at disease onset is variable even within pedigrees harbouring the same mutation. The late age at onset suggests a multistep process in which genetic factors are penetrant only when combined with lifestyle or environmental factors37. In addition, incomplete heritability of known pathogenic mutations implicates an interaction with environmental factors. To date, the only confirmed epidemiological risk factors associated with the development of ALS are age and male gender1. Recent Mendelian randomization studies38 have produced relatively robust evidence for a causal link between strenuous physical exercise39 and hyperlipidaemia40, and the risk of ALS. Other potential environmental risk factors proposed include smoking, military service and specific sporting activities, including soccer and American football41,42,43,44.
Box 2 Key pathophysiological insights from recent ALS genetic studies 1. Evidence for a role of oligogenic inheritance in which more than one gene causes the phenotype226,267,268, and also genetic pleiotropy in which mutations in a single gene may cause multiple phenotypes269. 2. Mutations in RNA processing/RNA binding proteins including C9orf72, FUS, TARDBP, ATXN2, SETX, ANG, hnRNPA1 and MATR3 are significantly associated with ALS. TDP-43 is a key protein in the pathophysiology of amyotrophic lateral sclerosis (ALS), not only because mutations in the encoding TARDBP gene can cause ALS, but because >97% of patients with ALS display TDP-43 proteinopathy. TDP-43 regulates RNA splicing, but also has a specific role in the splicing of cryptic exons270. Cryptic exon inclusion may interfere with the function of genes crucial for motor neuron health including UNC13A105 and STMN2 (ref. 271). 3. ALS-associated risk variants can be associated with other neurodegenerative phenotypes, most notably frontotemporal dementia (FTD). At least 13 genes have now been identified which can cause ALS, FTD or both conditions including C9orf72, CHCHD10, SQSTM1, TBK1, CCNF, FUS, TARDBP, OPTN, UBQLN2, TUBA4A, ATAXN2, VCP and CHMP2B32. 4. Several new ALS risk genes have been identified in recent years including ATXN2, CAV1, SPTLC1, ACSL5, DNAJC7, ANAXA11 and GLT8D272,273,274,275,276,277,278,279,280. Some of these have highlighted new pathomechanisms including: DNAJC7, a heat-shock protein co-chaperone probably linked to intracellular protein homeostasis276; GLT8D1, a glycosyltransferase enzyme linked to ganglioside biosynthesis and neurotrophic factor signalling275. 5. Loss of function mutations have been found in certain genes including TBK1 (ref. 281), NEK1 (ref. 282) and FIG4 (ref. 283). A gene replacement strategy could be a promising avenue for therapy advancement in these rare genetic subtypes of ALS, similar to survival motor neuron (SMN) replacement in spinal muscular atrophy284.
Oxidative stress
Oxidative stress may result from excess levels of reactive oxygen species (ROS), reactive nitrogen species or impaired functioning of antioxidant defence systems. ROS contribute significantly to neuronal injury and central nervous system (CNS) ageing by changing the structure and function of biomolecules including proteins, lipids, DNA and RNA. There is abundant evidence indicating a key role for oxidative damage in the pathophysiology of ALS45,46,47. Altered oxidative stress biomarker profiles have been reported in models of ALS and in human ALS biosamples48. There is also evidence for impaired oxidative stress defence systems in ALS, including dysregulation of glutathione homeostasis49 and the nuclear factor erythroid 2-related factor 2 (Nrf2) antioxidant response element (ARE) cytoprotective system50,51.
Oxidative stress can contribute to and exacerbate multiple other pathophysiological processes that are involved in motor neuron injury45. Biochemical and cell-based assays have identified oxidative stress as a signalling cue which promotes acetylated TDP-43 aggregates, while TDP-43 acetylation impairs RNA binding and promotes accumulation of insoluble, hyperphosphorylated TDP-43 species that resemble the pathological inclusions found in the CNS of individuals with ALS52. Mislocalization of both TDP-43 and FUS occur in cellular models of ALS exposed to oxidative stress, underpinning alterations in RNA processing53. Chronic oxidative stress promotes GADD34-mediated phosphorylation of TDP-43, a hallmark of TDP-43 proteinopathy54. Cytoplasmic aggregation of TDP-43 sequesters specific microRNAs and proteins, including nuclear genome-encoded mitochondrial proteins, leading to dysregulation of mitochondrial function that further augments oxidative stress55. Thus, an accelerating cycle of oxidative stress, protein aggregation and mitochondrial dysfunction is established.
Oxidative stress is also involved in the crosstalk between motor neurons and neighbouring astrocytes and microglia56,57. For example, there is evidence that astrocytes release glutamate via upregulation of the cysteine–glutamate antiporter in response to increased oxidative stress and this in turn may augment excitotoxic injury to motor neurons56.
Effective alleviation of oxidative stress could potentially ameliorate multiple facets of the pathobiology of motor neuron degeneration. A retrospective analysis of drugs evaluated in mSOD1 mice indicated that drugs that specifically target oxidative stress could be the most promising therapeutic candidates for the prevention of motor neuron degeneration58. Alleviation of oxidative stress is likely to be the primary mechanism of action of the free radical scavenging drug edaravone.
Excitotoxicity
Excitotoxicity mediated by excessive stimulation of postsynaptic glutamate receptors is considered a major pathophysiological mechanism in ALS59. A prolonged increase of synaptic glutamate causes excessive neuronal firing, increasing intracellular calcium levels with downstream neurotoxic effects60. Excitotoxicity causes prolonged pathological changes, such as ER stress and mitochondrial calcium overload56. Excessive glutamate exposure also decreases cysteine uptake by inhibiting the glutamate–cysteine antiporter, depleting the intracellular levels of the antioxidant glutathione and increasing indices of intracellular oxidative stress61.
Motor neurons susceptible to neurodegeneration in ALS have cell-specific features which increase their vulnerability to excitotoxic injury including: high expression of calcium-permeable AMPA receptors lacking the GluR2 subunit62 and low expression of the calcium-buffering proteins parvalbumin and calbindin63. Decreased expression of the major glutamate re-uptake transporter GLT1–EAAT2 has been reported in both animal models and the CNS of patients with ALS64,65,66. In human induced pluripotent stem cell (iPSC)-derived motor neurons, C9orf72 mutations increase Ca2+-permeable AMPA receptor-mediated excitotoxicity67 and also impair mitochondrial calcium-buffering capacity68.
Transcranial magnetic stimulation with threshold tracking has been deployed to demonstrate that cortical hyperexcitability is an early and intrinsic feature of sALS and fALS69. Magnetic resonance spectroscopy (MRS) has been used to measure glutamate and GABA levels in the brain of patients with ALS, with variable results across different studies.
Riluzole has complex and incompletely understood mechanisms of action. However, one component is an effect on presynaptic sodium channels that causes a reduction in the release of glutamate from presynaptic terminals, which is considered to ameliorate excitotoxic effects on motor neurons70. It is noteworthy that, in the current development pipeline, most active drug programmes targeting glutamate excitotoxicity are associated with riluzole reformulation efforts (for example, to produce stable liquid and sublingual preparations of the drug71). Metabotropic glutamate receptors are emerging as novel potential drug targets in ALS as their modulation may result in a decrease in glutamate release as well as induction of the production of neurotrophic factors (NTFs)72.
Mitochondrial dysfunction
Mitochondrial dysfunction has been proposed as a central determinant of the pathophysiology of ALS. Altered energy production, excess generation of ROS, disruption of mitochondrial axonal transport, altered mitochondrial structure and dynamics, perturbation of mitophagy and calcium buffering, and triggering of apoptosis have been extensively described in ALS model systems and patient biosamples73,74,75. Defects in the specialized domains on the ER mitochondrial-associated membranes have been reported to impair neuronal calcium homeostasis, mitochondrial dynamics, ER function and autophagy, culminating in axonal degeneration76.
Mutations in specific ALS genes have been linked to compromise of mitochondrial function through various mechanisms77 and multiple abnormal ALS disease-associated proteins have been shown to interact directly with mitochondria, impairing their function. For example, mutant SOD1 aggregates in the mitochondrial intermembrane space and reduces the activity of the electron transport chain (ETC) complexes. The DPR poly-GR produced in C9orf72-mutant ALS binds to the mitochondrial complex V component ATP5A1 and enhances its ubiquitination and degradation78. The C9ORF72 protein localizes to the inner mitochondrial membrane and regulates cellular energy homeostasis by stabilizing the translocase of inner mitochondrial domain containing 1 (TIMMDC1) protein, a crucial factor for the correct assembly of the oxidative phosphorylation complex. The function of mitochondrial complex 1 is impaired in patient-derived neurons from patients with C9orf72-mutant ALS79.
There is evidence that TDP-43 has a role in maintaining mitochondrial homeostasis by regulating the processing of mitochondrial transcripts, a function that is perturbed in the presence of mutations80. TDP-43 aggregates also sequester specific microRNAs and proteins that are nuclear genome-encoded mitochondrial proteins, and abnormal levels of expression cause dysregulation of mitochondrial function which in turn augments oxidative stress55.
Potential therapeutic strategies targeting mitochondrial biology have been attempted. Several drugs targeting mitochondrial function and/or ROS such as coenzyme Q10, dexpramipexole, olesoxime and creatine all showed promise in preclinical models, but were unsuccessful in clinical trials81,82,83,84. Tauroursodeoxycholic acid (TUDCA) modulates the mitochondrial pathway of neuronal injury by reducing ROS formation, inhibiting BAX translocation, cytochrome c release and caspase 3 activation85. A recently published phase II clinical trial involving 137 patients with ALS evaluated treatment with TUDCA combined with sodium phenylbutyrate over a 6-month period86,87 and showed evidence of slowing of the rate of decline on the ALS functional rating scale–revised (ALSFRS-R) score and increased survival. A European trial of TUDCA alone is ongoing (EudraCT 2018-002722-22). A recent preclinical study, using models harbouring SOD1 and TARDBP mutations, demonstrated that inhibition of protein phosphatase 1 prevented mitochondrial fragmentation, ETC impairment, disruption of axonal transport and cell death88 and this pathway is worth exploring further. 31P-MRS has been used to provide evidence for bioenergetic dysfunction in the brain and muscle of patients with ALS and provides a potential biomarker tool for use in clinical trials targeting bioenergetic dysfunction89.
Impaired protein homeostasis
The correct balance between protein production and degradation is controlled by a complex network involving the synthesis, folding, trafficking and degradation of proteins which respond to stress signals such as the unfolded protein responses of the ER and mitochondria and the cytosolic heat shock response. Loss of proteome fidelity, accumulation of non-native protein aggregates and oxidatively damaged protein species are key features of an ageing cell and may contribute to age-related neurodegenerative disorders90. There is extensive evidence that dysfunctional proteostasis contributes to the pathophysiology of ALS91,92. Several proteins encoded by genes causative in ALS regulate the proteostasis network directly or indirectly (Table 1) and some ALS-linked proteins (for example, SOD1 and TDP-43) are substrates for these pathways. C9ORF72, sequestosome 1/P62, optineurin and ubiquilin 2 play key roles in the initiation of autophagy; ubiquilin 2, alsin, FIG4, VCP and CHMP2B are important in the control of maturation of autophagosomes; TBK1 loss of function impairs substrate delivery to autophagosomes93,94,95,96,97,98,99.
Motor neurons may be particularly vulnerable to proteome stress based on their size and axonal arbour, the relatively low expression of ubiquitin proteasome genes in motor neurons that are vulnerable to degeneration in ALS100, and their relative inability to mount an effective heat shock response101.
Intracellular protein aggregates are a hallmark pathological feature of ALS. Studies focusing on the effects of TDP-43 mislocalization have highlighted both gain and loss of function consequences including altered regulation of splicing, hyperresponsiveness to cellular stress, increased DNA damage, widespread alterations in the transcriptome102,103 and alteration in the transport of ribosomal protein mRNAs to regulate local axonal translation104. TDP-43 has a key role as a repressor of cryptic exon inclusion during RNA splicing, and loss of TDP-43 from the nucleus leads to inclusion of a cryptic exon in UNC13A mRNA and reduced UNC13A protein expression, which may interfere with vesicle maturation during exocytosis and neurotransmitter release105. In mouse models, TDP-43 mislocalization has been shown to cause abnormal synaptic function and a hyperexcitability phenotype in the motor cortex106.
Stress granules (SGs) are dynamic membraneless compartments composed of mRNAs and RNA-binding proteins (RBPs) that assemble on a temporary basis to allow the cell to respond to stress by halting translation of the majority of mRNAs and directing the translation of cytoprotective proteins which allow the cell to mount an effective stress response. Normally SGs are highly dynamic structures, but in the presence of age-related changes or severe cellular stress, they may form solid aggregated inclusion bodies. SGs in ALS contain multiple RNAs and proteins and also proteins such as TDP-43 and FUS which possess so-called low complexity prion-like domains that are prone to aggregation107. There is a prevalent view that impaired SG disassembly facilitates the transition from a liquid to a solid phase, driving the formation of cytoplasmic TDP-43 inclusions108. Multiple ALS genes encode proteins which interfere with SG dynamics through mutations of their low-complexity domain including: TARDBP, FUS, EWSR1, TAF15, hnRNPA1, hnRNAPA2B1, ATXN2 and TIA1 (ref. 109). ANXA11mutations have been shown to cause dysregulation of SG disassembly110 and VCP mutations impair autophagy-dependent SG degradation and also autophagy-independent disassembly111.
Restoration of proteostasis to reduce the burden of aggregated proteins is regarded as an attractive therapeutic approach for neuroprotection. Pathways targeted in experimental models have included: upregulation of chaperones; manipulation of the heat shock response; protection from ER stress; maintenance of EiF2A (eukaryotic initiation factor 2A) in the active state; induction of autophagy; and activation of the proteasome machinery90. An array of small molecules have been identified which can modulate several aspects of TDP-43 proteinopathy including the level of expression, nucleocytoplasmic localization, cleavage and phosphorylation status of TDP-43 (ref. 112). The heat-shock inducer arimoclomol showed promising results preclinically113, but recently failed to show efficacy in clinical trials114. The therapeutic potential of autophagy induction has been explored in experimental models91. Some success was reported with rapamycin and trehalose, and fluphenazine, methotrimeprazine and berberine have emerged as hits from screening studies115,116,117.
Studies to identify small molecules that modulate the recruitment of TDP-43 and other RBPs into SGs have highlighted compounds with planar aromatic moieties in cellular models of ALS118. Targeting the VCP–FAF2 axis to enhance SG disassembly has also been proposed as a potential therapeutic approach to promote the clearance of persistent SGs, although this approach has not yet reached human trials119.
Neuroinflammation and glial contribution
Neuroinflammation is a pathological hallmark evident in preclinical models and the CNS of patients with ALS both histologically and in imaging studies120.
Astrocytes maintain the integrity of the blood–brain barrier and modulate inflammatory signalling through the release of pro-inflammatory cytokines (interleukin (IL)-1, IL-6 and tumour necrosis factor (TNF)) or anti-inflammatory molecules (prostaglandin E2 and transforming growth factor (TGF)-β)121,122,123. Astrocytes derived from fibroblasts from patients with ALS are toxic to cocultured motor neurons124. The exact mechanisms of this toxicity have not yet been established although impaired bioenergetic support through lactate release, and pro-nerve growth factor–p75 receptor signalling, may contribute125.
Microglia can adopt a toxic M1 phenotype or a neuroprotective M2 phenotype126,127,128. In mutant SOD1-transgenic mice, the microglia switched from the M2 to the M1 phenotype after disease onset129. Peripheral blood monocytes from patients with ALS are more readily activated and differentiated to a pro-inflammatory M1 phenotype, and represent a potential target for immunomodulatory therapy130.
Microglial NLRP3 inflammasome activation has been reported as a key contributor to neuroinflammation in ALS. NLRP3 inhibition could potentially become a therapeutic target to ameliorate microglia-driven neuroinflammation and disease progression in ALS57. The nuclear factor-kappa β (NF-κB) protein has been reported as a master regulator of inflammation in ALS131. NF-κB signalling was activated within glia during disease progression in mutant SOD1 mice. Notably, NF-κB signalling has been proposed to regulate microglial activation in ALS, as the localization of NF-κB activity and deletion of NF-κB signalling in microglia rescued motor neurons from microglia-mediated death and extended survival in ALS mice by impairing pro-inflammatory microglial activation132.
The responses of glia to TDP-43 pathology have been studied133,134,135. TDP-43 proteinopathy has been reported to cause inflammation by triggering cytoplasmic release of mitochondrial DNA which activates the cytoplasmic DNA-sensing cyclic GMP-AMP synthase (cGAS)–stimulator of interferon genes (STING) pathway. It is possible that inhibition of cGAS–STING could mitigate inflammatory pathology in ALS136,137.
Transcriptomic analysis of the motor cortex identified 1,573 differentially expressed inflammatory genes in sALS and indicated specific inflammatory molecular signatures for different patient subgroups, with the potential for personalized therapeutic targeting138. The C9ORF72 protein may modulate inflammation139 and there is evidence that reduced C9ORF72 levels cannot suppress inflammation mediated by the STING pathway140.
Biochemical and imaging biomarkers of glial activation have the potential to be used as pharmacodynamic indices in clinical trials. Patients with ALS exhibit higher cerebrospinal fluid (CSF) levels of chitotriosidase (Chit-1) and chitinase-3-like protein 1 (CHI3L1) than neurological disease controls and healthy controls, which correlate with the rate of disease progression121. Glial activation in vivo has been measured using translocator protein (TSPO) PET imaging and dynamic 18F-DPA714 or 11C-PBR28 PET–MRI141.
DNA damage and repair
Postmitotic cells such as neurons are highly susceptible to DNA damage and if unrepaired, cell death ensues136. DNA damage has been demonstrated in pathological studies showing elevated oxidized deoxyguanosine (OdG) in CNS tissue and biofluids from patients with ALS142,143. Recent evidence has been reported of elevated apurinic/apyrimidinic DNA sites (loss of purine/pyrimidine base from the DNA strand) and activation of the DNA damage response (DDR) in ALS144.
Several genes mutated in ALS lead to elevated DNA damage or have roles in DNA repair. C9orf72-related ALS shows elevated staining for γH2AX, a phosphorylated histone that acts as an important regulator of the DDR, in spinal motor neurons145. iPSC-derived motor neurons from patients with C9orf72-related ALS show a similar elevation in γH2AX which is partially rescued by inhibition of oxidative stress146. C9orf72 hexanucleotide expansions are prone to formation of G-quadruplex DNA structures147 which promote the formation of R-loops148. These DNA–RNA hybrid structures are substrates for DNA strand breaks, and have been detected in spinal cord tissue from patients with C9orf72-related ALS and mouse models in association with defective ataxia telangiectasia mutated (ATM)-mediated DNA repair signalling148.
Recently TDP-43 has been shown to play a role in DDR signalling via association with DDR proteins103. Loss of nuclear TDP-43 was also shown to correlate with strand breaks and DDR response in spinal cord tissue from patients with sALS103. Further in vitro assessment in cell models of DNA damage showed that TDP-43 participates in non-homologous end-joining-mediated repair. TARDBP mutations prevent this activity and lead to DNA damage, and DNA damage itself can cause TDP-43 mislocalization149. Loss of TDP-43 also increases R-loop formation and genome instability and the TARDBP mutation causes the same abnormalities150. FUS and NEK1 mutations have also been implicated in dysregulation of DNA repair151,152.
The emerging picture is of DNA damage and impaired repair being a common pathological feature. This is supported by multiple lines of evidence implicating loss of DDR or DNA repair functions of ALS-related genes. A causal link for DNA damage in motor neuron loss is not yet supported by the available evidence, but therapeutic opportunities to enhance repair or mitigate damage (for example, by amelioration of oxidative stress) are worth further investigation.
Dysregulated RNA metabolism and nucleocytoplasmic transport (NCT)
Disrupted RNA metabolism appears to play a key role in the pathophysiology of ALS. Both TDP-43 and FUS are RNA-binding proteins (RBPs) with major roles described in multiple aspects of RNA biology including splicing, transcription, stability, export and microRNA biogenesis153. Both proteins have binding sites for over 5,000 genes and regulate expression levels of hundreds of mRNAs. The most profound mutation-associated changes in expression occur in genes with very long introns and roles in synaptic function154,155. C9orf72 mutations lead to the accumulation of RNA foci which sequester a range of RBPs including TDP-43 and RNA export factors155,156. This sequestration has inevitable downstream effects on RNA metabolism149 including dysregulated splicing. Targeting the repeat expansion in C9orf72with antisense oligonucleotides (ASOs) was shown to prevent the formation of RNA foci and downstream effects157,158 and is under clinical investigation (Table 2). The sequestration of RBPs to C9orf72 RNA foci also licenses them for nuclear export, allowing the pathological transcripts to escape to the cytoplasmic ribosomal machinery, resulting in the production of toxic DPRs159. This has become a potential target for therapeutic intervention by inhibition of the nuclear export adaptor SRSF1 (ref. 159).
| Drug (company) | Mechanism of action | Development stage | Trial identifiers |
|---|---|---|---|
| Antioxidants | |||
| Verdiperstat (Biohaven Pharmaceuticals) | Myeloperoxidase inhibitor | Phase II/IIIa,b | NCT04297683, NCT04436510 |
| RT001 (Retrotope) | Provides resistance to membrane lipid peroxidation | Phase II | NCT04762589 |
| AP-101 (AL-S Pharma) | Anti-SOD1 monoclonal antibody | Phase II | NCT05039099 |
| Cell therapy | |||
| Lenzumestrocel (Corestem) | Mesenchymal stem cells that express anti-inflammatory and immune-modulating factors | Approved in South Korea | NCT04745299 |
| NurOwn (BrainStorm Cell Therapeutics) | Mesenchymal stem cells differentiated into astrocyte-like cells that express neurotrophic factors | Phase III (completed)c | NCT03280056, NCT04681118 |
| AstroRx (Kadimastem) | Pluripotent stem cell therapy | Phase I/II (completed) | NCT03482050 |
| RAPA-501 (Rapa Therapeutics) | Autologous hybrid T cell therapy (regulatory T cells and helper T cells) | Phase I/II | NCT04220190 |
| AlloRx (Vitro Biopharma) | Umbilical cord-derived allogeneic mesenchymal stem cell therapy | Phase I | NCT05003921 |
| Genetic therapy | |||
| Tofersen (Biogen, Ionis Pharmaceuticals) | Antisense oligonucleotide against SOD1 | Phase III (completed)c NDA submitted | NCT02623699 |
| ION-363 (Ionis Pharmaceuticals) | Antisense oligonucleotide against FUS | Phase III | NCT04768972 |
| WVE-004 (WaVe Life Sciences) | Antisense oligonucleotide against C9orf72 | Phase I/II | NCT04931862 |
| ION-541 (Ionis Pharmaceuticals) | Antisense oligonucleotide against Ataxin2 | Phase I | NCT04494256 |
| Mitochondrial dysfunction | |||
| AMX0035 (Amylyx Pharmaceuticals) | Combination of sodium phenylbutyrate and taurursodiol that reduces nerve cell death | Approved in the USA and Canada | NCT03127514, NCT05021536 |
| SBT-272 (Stealth BioTherapeutics) | Mitochondria-targeted novel peptide and peptidomimetic | Phase I | NA |
| Neuroinflammation | |||
| Masitinib (AB Science) | CSF1R kinase inhibitor | Phase III | NCT03127267 |
| Ibudilast (MediciNova) | Phosphodiesterase inhibitor | Phase II/III | NCT04057898 |
| 3K3A-APC (ZZ Biotech) | Recombinant engineered activated protein C | Phase II | NCT05039268 |
| Aldesleukin (Clinigen, Novartis) | Recombinant human IL-2 | Phase II/III (completed) | NCT02059759 |
| ANX-005 (Annexon) | C1q-specific mAb | Phase II | NCT04569435 |
| Apilimod dimesylate (AI Therapeutics) | Phosphatidylinositol-3-phosphate 5-kinase type III (PIKfyve) kinase inhibitor | Phase II | NCT05163886 |
| Tegoprubart (Eledon Pharmaceuticals) | Anti-CD40 ligand mAb that targets CD4+ T cells and B cells | Phase II (completed) | NCT04322149 |
| BLZ-945 (Novartis) | CSF1R inhibitor | Phase II | NCT04066244 |
| COYA-101 (Coya Therapeutics) | Autologous regulatory T cell therapy | Phase II | NCT04055623 |
| Fasudil (Woolsey Pharmaceuticals) | Rho kinase (ROCK) inhibitor | Phase II | NCT05218668 |
| Latozinemab (Alector) | Microglia-activating recombinant human anti-sortilin (SORT1) mAb | Phase II | NCT05053035 |
| Pegcetacoplan (Apellis Pharmaceuticals) | Complement C3 regulator | Phase II | NCT04579666 |
| PrimeC (NeuroSense Therapeutics) | Combination of celecoxib (cyclooxygenase-2 inhibitor) and ciprofloxacin (DNA gyrase inhibitor) | Phase II | NCT04165850, NCT05357950 |
| RNS60 (Revalesio) | Neuroprotective G protein-coupled receptor modulator | Phase II (completed) | NCT03456882 |
| CORT-113176 (Corcept Therapeutics) | Glucocorticoid receptor antagonist | Phase I (completed) | NCT04994743 |
| DNL-788 (Denali) | RIPK1 inhibitor | Phase I (completed) | NCT04982991 |
| Proteostasis | |||
| Trehalose (Seelos Therapeutics) | Repurposed disaccharide that may prevent mutant protein aggregation | Phase II/IIIa | NCT04297683, NCT05136885 |
| IFB-088 (InFlectis BioScience) | Inhibitor of PPP1RlsA (GADD34), involved in the unfolded protein response | Phase II | EudraCT 2021-003875-32 |
| Trametinib (GENUV) | Mitogen-activated protein kinase (MEK) inhibitor that aims to activate the autophagy–lysosome pathway | Phase I/II | NCT04326283 |
| ABBV-CLS-7262 (AbbVie/Calico) | eIF2B activator that inhibits the integrated stress response pathway | Phase I | NCT04948645 |
| DNL-343 (Denali Therapeutics) | eIF2B activator that targets integrated stress response pathway and others | Phase I | NCT05006352 |
| Monepantel (PharmAust) | Inhibits a signalling system controlled by mTOR and reduces protein accumulation | Phase I | NCT04894240 |
| Miscellaneous | |||
| Reldesemtiv (Cytokinetics) | Troponin activator | Phase III | NCT04944784 |
| Triumeq (Neuroscience Trials Australia) | Combination of abacavir, dolutegravir and lamivudine that inhibits human endogenous retroviruses (HERV) | Phase III | NCT02868580, EudraCT 2020-005069-15 |
| Pridopidine (Prilenia Therapeutics) | Dopamine and σ1 receptor agonist that increases production of brain-derived neurotrophic factor | Phase II/IIIa | NCT04297683, NCT04615923 |
| CNM-Au8 (Clene Nanomedicine) | A gold nanocrystal suspension that aims to enhance cellular bioenergetics | Phase II/IIIa | NCT04297683, NCT04414345 |
| ILB (TikoMed) | Low molecular weight dextran sulfate with multiple actions | Phase II (completed) | NCT03705390 |
| PTC-857 (BioElectron Technology) | 15-Lipoxygenase inhibitor | Phase II | NCT05349721 |
| Donaperminogene seltoplasmid (Helixmith) | DNA plasmid that expresses recombinant hepatocyte growth factor to promote neuroregeneration | Phase II | NCT04632225 |
| TPN-101 (Transposon Therapeutics) | Nucleoside analogue reverse transcriptase inhibitor | Phase II | NCT04993755 |
| BIIB100 (Biogen) | Exportin 1 inhibitor that targets nucleocytoplasmic transport | Development discontinued after phase I | NCT03945279 |
| mEphA4-Fc (NuNerve) | Competitive inhibitor of EphA4 protein that promotes recovery of motor neurons following injury | Phase I | ACTRN 12621000514808 (Australia) |
| Prosetin (ProJenX) | Microtubule-associated protein 4 kinase inhibitor that aims to target glutamate excitotoxicity | Phase I | NCT05279755 |
Selected drugs in clinical trials for ALS
Drugs under clinical development for the treatment of ALS were identified using the AdisInsight Springer database (August 2022). Amongst these, selected active drug development programmes with at least one significant ALS trial updated within the previous 12 months are listed in the table. ALS, amyotrophic lateral sclerosis; CSF1R, colony-stimulating factor 1 receptor; eIF2b, eukaryotic translation initiation factor 2b; IL, interleukin; mAb, monoclonal antibody; mTOR, mammalian target of rapamycin; NA, not applicable; NDA, new drug application. aThese trials are part of the HEALEY ALS Platform trial. bThe primary end point was not achieved. cThe primary end point was not achieved but positive effects on biomarkers were found.
Deficiencies in NCT have been identified in multiple ALS model systems160,161,162,163. DPRs resulting from repeat-associated non-AUG (RAN) translation of C9orf72 repeats promote SG formation107, and an emerging model is one in which aberrant recruitment of NCT factors to SGs disrupts NCT, leading to neurodegeneration164. Inhibitors of the integrated stress response (ISR) such as ISRIB, a stabilizer of eIF2B function165 were able to protect against these effects164 and related compounds are in clinical studies in ALS (Table 2).
The mislocalization of TDP-43 itself is thought to play a role in disturbed NCT via sequestration of NCT machinery components166. A putative target to reduce TDP-43 nuclear export is exportin 1 which binds the nuclear export sequence in proteins to mediate their export from the nucleus. Inhibitors of exportin-1 (KPT-276 and KPT-335) have been shown to reduce cell death in cortical neurons expressing a TDP-43 carboxy-terminal fragment166. However, whether TDP-43 nuclear export is mediated by exportin 1 is still not established167. An ASO approach to limit exportin-1 expression (BIIB100) was discontinued after phase I trials.
Unidirectional transport of many cargoes is dictated by a cytoplasmic to nuclear gradient of RAN GTPase, which exists predominantly in a GTP-bound state in the nucleus. The loss of this gradient is predicted to be a key factor in the disruption of NCT in ALS. A pathway which maintains this gradient and restored normal NCT in C9orf72-related ALS neurons has recently been identified168. Overall, the pathways related to NCT may be fertile ground for future therapeutic discovery in ALS.
Impaired axonal transport and integrity
Due to their extreme polarization, motor neurons are particularly reliant on retrograde and anterograde transport of cargos to maintain axonal integrity. Defects in axonal transport have been described in multiple ALS models and in the human disease, as evidenced by pathological accumulations of organelles and phosphorylated neurofilaments in motor neuron axons, as well as the identification of ALS-associated mutations in cytoskeletal and axonal transport-related genes169. Mutations in KIF5A, which encodes a microtubule motor protein, and in ANXA11, which disrupt axonal RNA transport, were recently identified as causative in ALS170,171.
Targets which may be amenable to therapeutic intervention have been identified. Small-molecule screens identified P38 MAPK inhibitors that rescued axonal transport defects in SOD1-mutant mice172 and IGF1R inhibitors173 as selective potentiators of the axonal transport of signalling endosomes. Retrograde transport of neurotrophin-containing signalling endosomes is required for axonal integrity, and inhibition of P38 MAPK or IGF1R increased their rate of retrograde axonal transport172,173.
A well-described activator of axonal transport is via inhibition of HDAC6 and consequent reduction in tubulin acetylation. Knockdown or pharmacological inhibition of HDAC6 restored axonal transport defects in human FUS-associated ALS cellular models174. In vivo models have pointed to a prominent role of mutant TDP-43 in mediating axonal transport defects175. Compounds targeting HDAC6 are currently in the preclinical discovery phase for the treatment of ALS.
Wallerian degeneration is a process of controlled axonal degeneration involving enzymes which detect and trigger programmed axonal degeneration following injury176. Sterile-α and Toll/IL-1 receptor (TIR) motif-containing protein 1 (SARM1) is a NAD-degrading enzyme with a major role in facilitating fast Wallerian degeneration. Sarm1 deletion in a transgenic TARDBPQ331K mouse model reduced motor axon degeneration and denervation of neuromuscular junctions177. SARM1 inhibitors are currently being explored for several therapeutic indications, including ALS. Overall, the mechanisms underlying disrupted axonal transport and integrity are a promising avenue for future therapeutic discovery.
ALS drug pipeline
Pathophysiological mechanisms currently being targeted in preclinical development and clinical trials for ALS are highlighted in Fig. 2.
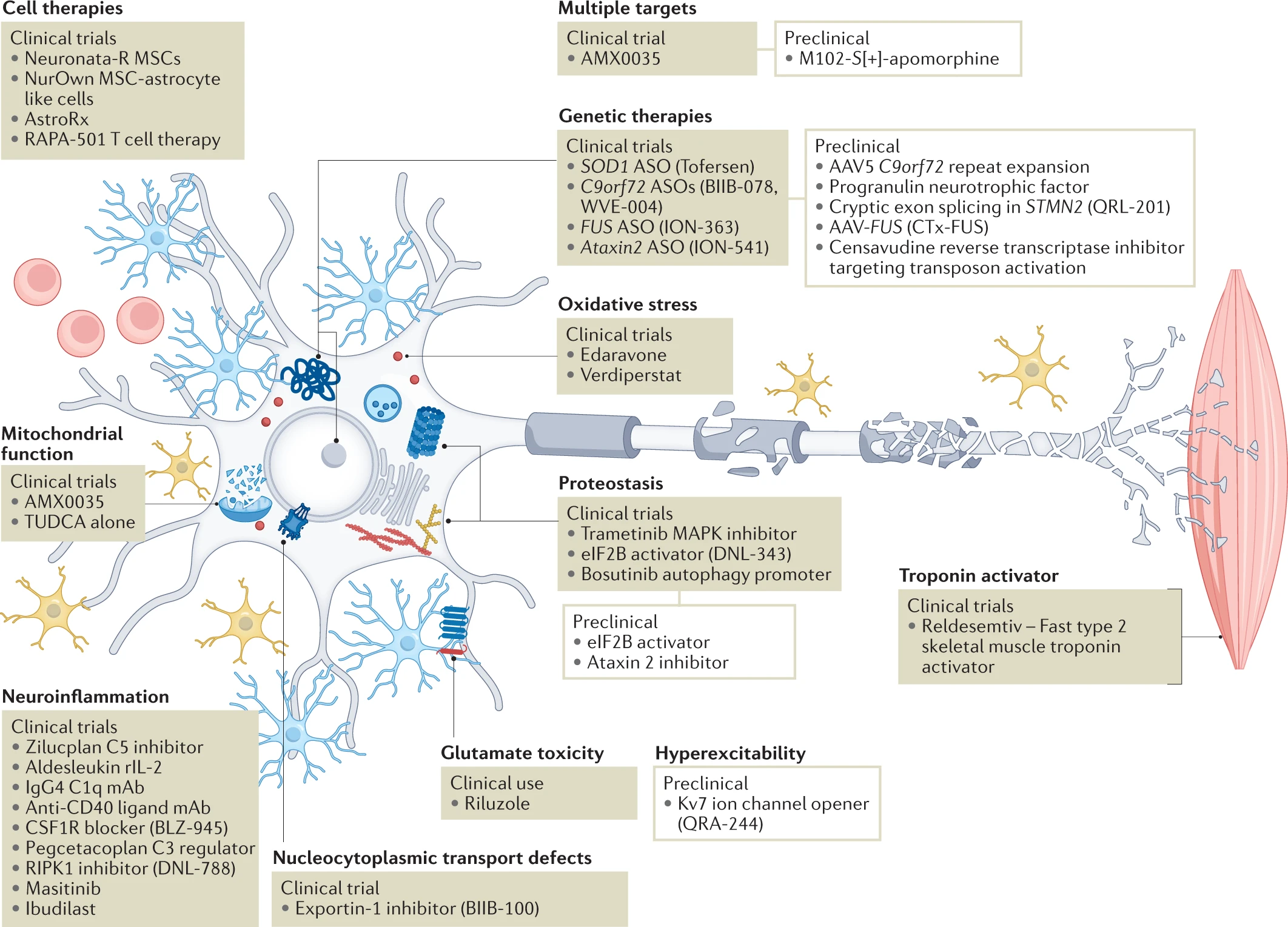
The major ALS pathophysiological targets currently being pursued.
We have identified over 40 active clinical programmes (Table 2) and over 50 late-stage preclinical programmes (Table 3) and show here how these efforts map onto the pathophysiological landscape of amyotrophic lateral sclerosis (ALS). Among clinical programmes, some trends emerge. Neuroinflammation is the best represented therapeutic target covered by a variety of approaches, with the complement system representing a particular target of interest. Genetic therapies using antisense oligonucleotide (ASO) approaches also dominate the clinical space targeting specific ALS genes, although Ataxin2modifiers may be more broadly applicable in relation to TDP-43 proteinopathy. Genetic therapies are also prominent in preclinical efforts. Other notable areas include proteostasis and cell therapies. Considering the most up to date understanding of pathomechanisms and genetics, several areas are poised for further exploration in sporadic ALS more broadly, including RNA metabolism and axonopathy. AAV, adeno‐associated virus; mAb, monoclonal antibody; MSC, mesenchymal stem cell; TUDCA, tauroursodeoxycholic acid.
We identified 91 development programmes for drugs approved or in clinical development for the treatment of ALS, using the AdisInsight Springer database (August 2022). A total of 53 drug development programmes had at least one significant ALS trial update within the last 12 months and were not discontinued programmes (Table 2). Of the 53 drug development programmes, 39 focused on novel therapeutic agents, while the other 14 were related to marketed drugs, drug repurposing or drug reformulation (including six drugs that are reformulations of edaravone or riluzole, not listed in Table 2). Six drugs were either marketed or in the pre-registration stage, while 11 additional drugs were under evaluation in pivotal trials.
The selected 53 drugs with recent updates were manually categorized according to the mechanism of action to identify trends and gaps, and several key themes emerged. The most represented target category was neuroinflammation, with 16 therapeutic agents in trials using approaches ranging from upregulation and activation of endogenous regulatory T cells (aldesleukin) to therapeutic antibodies targeting CD4+ T cells and B cells (anti-CD40L). Several approaches targeting the complement cascade, including ANX-005 (C1q inhibitor) and pegcetacoplan (C5 convertase inhibitor) were identified, although recent trials in ALS of the anti-C5 antibody ravulizumab and the C5 inhibitor zilucoplan were terminated following interim analyses, due to lack of efficacy.
Another well-represented group of compounds in trials was for genetic therapies using ASO technology to knockdown gene expression. Three of these target specific mutations in genetic subpopulations (C9orf72, FUS and SOD1) and one targets Ataxin2, a potential modifier of TDP-43 proteinopathy, which could be relevant for almost all ALS subtypes. Depletion of ataxin 2 levels limits TDP-43-mediated toxicity in multiple model systems178. The ASO ION-363 is currently being evaluated in a phase III trial (NCT04768972) targeting patients with ALS and mutations in the FUS gene. ION-363 was initially developed as an experimental treatment specifically for a patient with FUS-associated ALS and then provided to several patients in a compassionate use programme. The controlled phase III trial was established based on this initial programme179.
Other well-represented approaches include glutamate excitotoxicity, although the four different approaches identified represent generic bioequivalence trials for new formulations of riluzole. Cell therapy continues to be an area of interest, although the current focus is on cell therapy that modulates the toxic microenvironment, as opposed to cell replacement. NurOwn is one of the larger recent cell therapy trials, although the pivotal phase III trial (NCT03280056) did not meet statistical significance in the primary efficacy end point180. Additionally, troponin activators that increase muscle force for a given level of motor neuron innervation represent a symptomatic approach to improve muscle strength, rather than aiming for neuroprotection of motor neurons181.
Approaches that are under-represented, based on current knowledge of the pathophysiology of ALS, include those targeting mitochondrial dysfunction and proteostasis. AMX0035 potentially targets both of these pathways and was the subject of a recent new drug application (NDA) and marketing authorization application filings based on data from phase II trials86,87. AMX0035 was approved in 2022 for use in the USA and Canada for the treatment of ALS. The phase III PHOENIX trial of this combination therapy is ongoing.
Clinical case studies
Analysis of clinical stage ALS drug development programmes has highlighted several major target mechanisms of action. To demonstrate the current trends in ALS drug development, one representative drug programme from each target mechanism of action was selected for further discussion in the following case studies.
NurOwn
Mesenchymal stem cell (MSC)-NTF cell therapy (NurOwn), developed by BrainStorm Cell Therapeutics182, is the first ALS cell therapy programme evaluated by a randomized, placebo-controlled pivotal trial. Adult stem cells were collected from the patient’s own bone marrow, expanded and manipulated to secrete NTFs ex vivo. Subsequently, the cells were transplanted back into the patient via intrathecal injection. It was demonstrated that adult bone marrow MSCs may be differentiated into astrocyte-resembling cells that express astrocytic markers and produce significant levels of NTFs183,184. These MSC-NTF cells secrete NTFs, such as glial cell line-derived NTF (GDNF), brain-derived NTF (BDNF), vascular endothelial growth factor (VEGF), and hepatocyte growth factor (HGF) with the potential to protect motor neurons. MSC-NTF cells are also reported to stimulate new motor neuron growth and to promote neuromuscular junction reinnervation182.
A pivotal phase III trial (NCT03280056) was conducted to evaluate the safety and efficacy of repeated administration of NurOwn in patients with ALS. The randomized, double-blind trial completed enrolment of 196 patients. The trial did not meet statistical significance in the primary efficacy end point, which evaluated the percentage of trial participants who experienced a 1.25 points per month improvement in the post-treatment ALSFRS-R slope. Response rates of 33% and 28% were observed for the NurOwn and placebo groups, respectively (P = 0.45). However, positive elements did emerge in the phase III trial. For example, significant improvements in CSF biomarkers of neuroinflammation, neurodegeneration and NTF support were observed in patients treated with NurOwn, and the treatment was well tolerated87,180. Meanwhile, another MSC cell therapy programme, lenzumestrocel (Neuronata-R), is currently under evaluation in a phase III clinical trial (NCT04745299) to confirm efficacy and safety. Neuronata-R therapy was approved for the treatment of ALS by the Ministry of Food and Drug Safety in South Korea in 2014.
Tofersen
Tofersen is an ASO developed by Ionis Pharmaceuticals and licensed to Biogen, and is in development for the treatment of patients with ALS and a confirmed SOD1 mutation. Tofersen binds to SOD1 mRNA and is predicted to reduce the synthesis of the SOD1 protein and therefore ameliorate the toxic gain of function of mutant SOD1. Tofersen is administered by intrathecal injection at monthly intervals.
Tofersen was evaluated in SOD1–transgenic rodent models. The drug extended survival by more than 50 and 40 days in SOD1G93A rats and SOD1G93A mice, respectively185. The initial loss of compound muscle action potential in SOD1G93A mice was also reversed after a single dose of tofersen. Furthermore, increases in serum phosphorylated neurofilament heavy, a promising biomarker for ALS, were ameliorated by the ASO. Confidence in the approach for SOD1 gene silencing also arose from gene therapy trials in SOD1-transgenic mice using intrathecal administration of adeno-associated viral vectors to deliver short hairpin RNAs targeting SOD1 (ref. 186).
In the reported phase I/II trial (NCT02623699) of tofersen, involving 50 participants with SOD1-associated ALS, the mean level of CSF SOD1 was lowered by 33% on day 85 in the group receiving the highest dose (100 mg) compared with the group receiving placebo. The concentrations of phosphorylated neurofilament heavy and neurofilament light (NFL) in plasma and CSF were decreased during the intervention period among the ten participants who received 100 mg of tofersen, providing the first indication in an ALS trial of a potential biomarker of therapeutic efficacy. Additional exploratory measures suggested that tofersen may slow the functional decline of patients, as assessed by ALSFRS-R, pulmonary function testing and evaluation of muscle strength by hand-held dynamometry187.
Based on the above positive outcome, an additional cohort was recruited to evaluate the 100 mg dose in the tofersen phase III VALOR trial, which enrolled 108 patients internationally and completed in 2021. The VALOR trial did not achieve statistical significance on its primary end point of change in ALSFRS-R score at 6 months. However, consistent positive effects were observed across key secondary and exploratory clinical outcome measures, and these effects became more apparent with longer-term follow-up in the open-label extension study after the 6-month experimental period. Tofersen administration resulted in a slowing of decline in participants with faster progressing disease, as well as an apparent clinical stabilization in participants with slower progressing disease188,189. Biogen has filed an NDA submission with the FDA, which accepted the application in July 2022 under the accelerated pathway, and granted priority review. In October 2022, Biogen announced that the FDA had extended the review period of the NDA for tofersen by 3 months. Therefore, the updated Prescription Drug User Fee Act goal date is 25 April 2023. Biogen has also initiated a phase III ATLAS study to evaluate the ability of tofersen to delay clinical onset of ALS in pre-symptomatic individuals with a SOD1 mutation and biomarker evidence of ALS disease activity measured by a threshold level of NFL in plasma (NCT04856982). This will allow the effects of early intervention to be evaluated.
A similar approach, using intrathecally delivered ASOs, has been employed for the treatment of C9orf72-associated ALS (BIIB078 (terminated in 2022 due to lack of efficacy) and WVE004). During these trials, the measurement of DPRs in CSF could provide a useful biomarker of target engagement190. The BIIB078 ASO led to robust dose-dependent reduction of DPRs in the CSF. However, despite this, the treatment group receiving the highest dose showed a trend towards greater decline than the group receiving placebo across clinical outcome measures as well as an increase in NFL levels, suggesting increased neuronal injury191. The reasons for this unexpected outcome are uncertain but could include: the fact that the ASO targeted only the sense transcripts produced by the C9orf72 expansion, leaving antisense transcripts untouched; failure to address or possible exacerbation of C9 haploinsufficiency; or failure to adequately address downstream mechanisms of disease propagation (for example, TDP-43 proteinopathy).
AMX0035
AMX0035 (Relyvrio) is the first combination potential disease-modifying therapy trialled in ALS, although another combination therapy of dextromethorphan hydrobromide and quinidine sulfate was approved for the symptomatic treatment of pseudobulbar affect in ALS (NCT00573443, NCT01799941; Nuedexta Prescribing Information). AMX0035 is an oral proprietary, fixed-dose combination of sodium phenylbutyrate and taurursodiol (also known as TUDCA) developed by Amylyx Pharmaceuticals as a therapeutic approach targeting multiple pathophysiological mechanisms11 in ALS and Alzheimer disease. Sodium phenylbutyrate is a histone deacetylase inhibitor that upregulates chaperones192, while taurursodiol inhibits mitochondrial associated apoptosis193.
In the phase II CENTAUR trial (NCT03127514), 137 participants were randomly assigned 2:1 to receive AMX0035 or placebo for 6 months. Participants completing the 6-month randomized phase were eligible to receive phenylbutyrate and taurursodiol in an open-label extension. In a modified intention-to-treat analysis, AMX0035 was associated with a statistically significantly lower mean rate of change in the ALSFRS-R score at −1.24 points per month, compared to −1.66 points per month in those receiving placebo. Secondary outcomes did not differ significantly between the two groups87. Median overall survival was 25.0 months in participants originally randomized to AMX0035 and 18.5 months in those originally randomized to placebo. Initiation of active treatment at baseline resulted in a 6.5-month longer median survival compared to placebo. These trial results suggest that AMX0035 may have functional and survival benefits in ALS86 although several aspects of the trial have been criticized194.
Amylyx has sought market authorization from multiple regulatory authorities and has also initiated recruitment into a phase III PHOENIX trial evaluating the safety and efficacy of AMX0035 in approximately 600 patients with ALS (NCT05021536). AMX0035 has received market authorization in the USA and Canada for the treatment of ALS (2022). The drug approval by Health Canada was under its notice of compliance with conditions policy, which enables therapies showing the potential to fill an unmet medical need in severe, life-threatening diseases to reach the market sooner, provided that certain conditions are met. Those conditions included the provision of data from the phase III Phoenix trial (NCT05021536), as well as additional planned or ongoing studies. Several post-marketing studies were also required by the FDA for the approval of AMX0035, including carcinogenicity studies, additional drug interaction studies, and clinical pharmacokinetic studies in subjects with hepatic and renal impairment195.
One of the active ingredients of AMX0035, taurursodiol, is currently being evaluated in a randomized phase III TUDCA-ALS trial as an add-on treatment to riluzole in patients with ALS, sponsored by Humanitas Mirasole SpA (NCT03800524).
Aldesleukin
Aldesleukin is being investigated in a phase II/III ALS clinical trial (MIROCALS), with an embedded experimental medicine approach. Aldesleukin, recombinant human IL-2 with essentially similar pharmacodynamic properties to endogenous IL-2, is being developed by Clinigen for the treatment of ALS. It has been approved by the FDA for the treatment of metastatic melanoma and renal cell carcinoma under the brand name Proleukin. In vitro studies using human cell lines demonstrated the immunoregulatory properties of aldesleukin, including activation and proliferation of cytotoxic T lymphocytes and natural killer and lymphokine-activated killer cells (Proleukin Prescribing Information). It was also noted that low-dose IL-2 enhanced regulatory T (Treg) cell function in autoinflammatory conditions, while neuroinflammation represents a key pathogenic feature of ALS196,197.
The phase II IMODALS trial (NCT02059759) was conducted to evaluate low-dose aldesleukin in 36 patients with ALS. The trial met its primary end point, with both doses of aldesleukin resulting in a significant, dose-dependent, increase in Treg cell numbers, compared with placebo. This increase was accompanied by enhancement of Treg cell suppressive function, suggesting an improved ability to control the inflammatory mechanisms that contribute to neuronal injury in ALS198. There was also a significant, dose-dependent drop in CCL2 (MCP-1) levels, an inflammatory biomarker which is frequently elevated in patients with ALS197. The peripheral blood mononuclear cell (PBMC) transcriptome was also analysed at different time points198. Participants were classified into low, moderate and high responders based on the magnitude of the upregulation in the Treg cell count. Substantial baseline differences were observed between the PBMC transcriptomes of participants, with the least responsive patients showing a more inflammatory-prone phenotype at baseline. Low-dose IL-2 caused a reduction in the pro-inflammatory transcriptome signature in both high-responder and low-responder groups. It was also observed that pretreatment expression levels of two genes correlated with the magnitude of drug responsiveness, allowing the development of a two-biomarker-based regression model to predict the Treg cell response to low-dose IL-2. These findings and the application of this embedded experimental medicine approach could be particularly relevant for future application of precision medicine approaches in ALS clinical trial design198.
The IMODALS trial data supported the evaluation of aldesleukin in the randomized phase II/III MIROCALS trial (NCT03039673) to further evaluate the safety and effectiveness of the most favourable dose (2 million IU/day subcutaneous injection for five consecutive days every 4 weeks) in a larger trial of 220 participants over a longer period of 18 months. The outcome of this trial is currently under analysis.
Ravulizumab
Several approaches targeting the complement cascade are currently in clinical trials for the treatment of ALS, and it may be informative to consider the recent failure of ravulizumab in the CHAMPION-ALS trial. Complement is an essential effector component of both innate and adaptive humoral immune responses activated by the alternative, lectin and classic pathways. It is well known that complement can be activated in response to CNS inflammation, enhancing tissue injury, and complement deposition has been identified in CNS tissue from patients with ALS199. A potential role for complement in both central motor neuron loss and peripheral neuromuscular junction injury200 has stimulated interest in targeting the complement cascade in ALS clinical trials.
Ravulizumab is a humanized anti-C5 antibody developed by Alexion and AstraZeneca for the treatment of ALS and other rare diseases. It is a terminal complement inhibitor that specifically binds to the C5 complement protein with high affinity, thereby inhibiting its cleavage to C5a and C5b, and preventing the generation of the terminal membrane attack complex C5b9 and production of the pro-inflammatory C5a fragment. Ravulizumab inhibits terminal complement-mediated intravascular haemolysis and has been approved in the USA for the treatment of paroxysmal nocturnal haemoglobinuria.
The phase III CHAMPION-ALS trial (NCT04248465) was conducted to evaluate the efficacy and safety of ravulizumab for the treatment of ALS. AstraZeneca discontinued the CHAMPION-ALS trial in August 2021 due to lack of efficacy, following a prespecified interim analysis. This outcome suggests that peripheral targeting of the complement terminal pathway is not beneficial in ALS. More recently, a phase II/III trial of another C5 inhibitor zilucoplan (NCT04297683, NCT04436497) was also discontinued for futility. However, further approaches target C3 convertases and utilize smaller peptidic inhibitors, which may have better access to the CNS and the potential to target an earlier phase in the complement activation pathway.
Verdiperstat
Verdiperstat is an orally administered small molecule inhibitor of myeloperoxidase (MPO) developed by Biohaven Pharmaceuticals. MPO increases oxidative stress and inflammation in the brain, which play roles in multiple neurodegenerative diseases. Inhibition of MPO could potentially alleviate the pathological mechanisms associated with neuroinflammation. A placebo-controlled phase II/III trial (NCT04297683, NCT04436510) under the HEALEY ALS Platform Trial was conducted in 167 patients to evaluate the safety and efficacy of verdiperstat. In September 2022, Biohaven reported the failure of this trial. Specifically, the drug did not achieve statistically significant differences on the prespecified primary outcome, disease progression, as measured by ALSFRS-R, survival and other key secondary measures. The safety profile of verdiperstat appeared to be similar to that found in previous clinical trials. More details of the study results are expected to be provided in the near future. The failure of verdiperstat highlights the complexity of patholophysiological mechanisms involved in ALS. It is possible that therapies targeting multiple pathways contributing to neuronal injury may have a higher probability of success.
Drugs in preclinical development
A search using the AdisInsight Springer database (August 2022) identified 91 preclinical ALS drug development programmes. Of these, 22 defined a lead therapeutic candidate, listed ALS as the lead target indication, had had at least one significant update within the previous 12 months, and were not discontinued programmes (Table 3). Moreover, all drug development programmes except one (a riluzole microencapsulated formulation) were focused on the development of new therapies. Of the preclinical development programmes, 11 were for genetic therapies, which represent an exciting new therapeutic approach for subgroups of patients with ALS.
| Drug (company) | Approach | Mechanism |
|---|---|---|
| QRL-201 (QurAlis Corporation) | Cryptic exon splicing | Aims to restore expression of STATHMIN-2 (STMN2), reduced due to TDP-43 pathology; STMN2 is a protein important for neural repair and axonal stability |
| Censavudine (Transposon Therapeutics) | Endogenous retrovirus | A nucleoside reverse transcriptase inhibitor aiming to inhibit transposon activation |
| GNK-301 (GeNeuro) | Endogenous retrovirus | An anti-human endogenous retrovirus (HERV-K) monoclonal antibody that blocks activity of the HERV-K envelope protein |
| ALN-SOD (Alnylam Pharmaceuticals) | Genetic therapy | An RNA interference therapeutic targeting SOD1 for treatment of SOD1-mutant ALS |
| ALPHA-0602 (Alpha Cognition) | Genetic therapy | Gene therapy that delivers progranulin, a neurotrophic protein regulating neuronal survival and inflammatory processes |
| AMT-161 (uniQure) | Genetic therapy | AAV5 gene therapy targeting repeat-expanded C9orf72 to lower toxic RNA aggregates and prevent dipeptide protein formation |
| AS-202 (AcuraStem) | Genetic therapy | PIKfyve-suppressing antisense oligonucleotide targeting TDP-43 pathology |
| BMD-001 (BIORCHESTRA) | Genetic therapy | Micro-RNA antisense oligonucleotide that regulates multiple mRNAs, reduces neuroinflammation and improves neurorestoration |
| Caveolin-1 gene therapy (Eikonoklastes Therapeutics) | Genetic therapy | Caveolin-1 is a membrane/lipid raft scaffolding protein that promotes neurotrophin effects and enhances neuroplasticity, mitochondrial health and neuronal adaptation to stress |
| CTx-FUS (Coave Therapeutics) | Genetic therapy | AAV gene therapy targeting FUS, a nuclear DNA/RNA binding protein that regulates various steps of gene expression |
| TW-002 (Treeway, Ferrer) | Genetic therapy | AAV gene therapy that upregulates glial cell line-derived neurotrophic factor |
| VTx-001 (VectorY Therapeutics) | Genetic therapy | AAV5-vectorized scFv that binds and neutralizes oxidized phospholipids |
| VTx-002 (VectorY Therapeutics) | Genetic therapy | AAV5-vectorized scFv that targets misfolded toxic TDP-43 species |
| QRA-244 (QurAlis Corporation) | Hyperexcitability | A Kv7 ion channel opener that controls motor neuron hyperexcitability-induced excitotoxicity |
| M102 (Aclipse Therapeutics) | Various | Nrf2 and HSF1 dual activator that engages multiple neuroprotective mechanisms |
| AGS-499 (Neuromagen Pharma) | Neurogenesis | Small-molecule telomerase reverse transcriptase that has neurogenic and neuroprotective effects |
| Alpha-5 (Pasithea Therapeutics) | Neuroinflammation | Anti-integrin mAb developed for the treatment of ALS and other neuroinflammatory disorders |
| COYA-201 (Coya Therapeutics) | Neuroinflammation | Treg cell-derived exosome therapy that promotes immunosuppressive and anti-inflammatory effects in cells and tissues |
| LP143 (LongBoard Pharmaceuticals) | Neuroinflammation | Cannabinoid receptor type 2 agonist, aiming to reduce microglial neuroinflammation |
| PMN-267 (ProMIS Neurosciences) | Proteostasis | mAb that targets the formation of misfolded TDP-43, which aggregates in neurons |
Selected drugs in preclinical development for the treatment of ALS
ALS drugs in preclinical development were identified using the AdisInsight Springer database (August 2022). Amongst these, selected active drug programmes with the lead target indication of ALS and at least one significant update within the previous 12 months are listed in the table. AAV, adeno‐associated virus; FUS, fused in sarcoma; HSF1, heat shock factor 1; mAb, monoclonal antibody; Nrf2, nuclear factor erythroid 2-related factor 2; scFV, single-chain variable fragment; SOD1, superoxide dismutase 1; TDP-43, TAR DNA-binding protein-43; Treg cell, regulatory T cell.
Preclinical drug development programmes are highlighted in Table 3 and themes emerge which indicate the potential future direction of ALS clinical trials. The best represented category is genetic therapy approaches targeting specific misfolded proteins (for example, TDP-43, FUS and SOD1). Neuroinflammation is less well represented preclinically, with three approaches documented (Alpha-5, COYA-201 and LP143). Several novel mechanisms are represented for the first time. Cortical hyperexcitability is an emerging mechanism, and QRA-244, which targets voltage-gated potassium channel Kv7 family members, represents a potential approach to address this pathophysiological mechanism. Preclinical and clinical studies highlighting the potential of inhibiting retrovirally-derived transposable elements from causing genomic instability has led to the strategy of repurposing existing CNS-penetrant reverse transcriptase inhibitors such as censavudine and Triumeq. HDAC-6 inhibitors for correction of axonal transport defects are noteworthy, as this mechanism contributes in multiple neurodegenerative diseases, although a lead compound has not yet been identified. Three representative preclinical drug development programmes are highlighted in the following case studies.
QRA-244
QRA-244 is a Kv7 ion channel opener developed by the QurAlis Corporation for the treatment of motor neuron hyperexcitability-induced disease progression in ALS. QurAlis utilized a live-cell screening strategy to investigate abnormal electrophysiological properties and reveal targets that modulate the intrinsic hyperexcitability of ALS motor neurons reprogrammed from fibroblasts201. This unbiased screen identified Kv7 as a strongly upregulated drug target. Kv7 as a therapeutic target was further supported by a recent clinical study of a Kv7 channel opener that decreased motor neuron excitability202. QurAlis conducted a precision medicine programme to develop QRA-244, a selective Kv7 ion channel opener, for the treatment of ALS. Preclinical development of QRA-244 is underway in the USA.
M102
M102 is an orally bioavailable small molecule under development by Aclipse Therapeutics in collaboration with the University of Sheffield, UK. M102 is the S-enantiomer of the marketed R-enantiomer Apokyn (R-apomorphine), a dopamine agonist used in the management of advanced Parkinson disease. The S-enantiomer is a very weak dopamine antagonist203,204, and thus M102 does not show the adverse effects associated with dopamine agonism that occur following exposure to R-apomorphine.
M102 was identified as a lead therapeutic candidate by the University of Sheffield in an effort to identify CNS-penetrant activators of the Nrf2 pathway205,206. M102 is unique as a pro-electrophilic drug which only activates the Nrf2 pathway in cells under oxidative stress, thus reducing potential off-target effects207,208. Importantly, M102 at similar doses is also capable of activating the heat shock factor 1 (HSF1) signalling pathway that upregulates molecular chaperones to improve proteotoxic stress209.
Preclinical pharmacological data demonstrated that M102 significantly slows disease progression in SOD1G93A mice205 and reversed disease indices in the TDP-43Q331K mouse model210. Unlike currently available ALS drugs, Nrf2 and HSF1 activation target multiple pathophysiological mechanisms contributing to motor neuron injury. M102 has received orphan drug designation from the FDA and EMA and is expected to advance to phase I/II clinical trials in the near future.
COYA-201
COYA-201 is a Treg cell-derived exosome preparation under development by Coya Therapeutics for the treatment of ALS. Neuroinflammation contributes to neurodegeneration, which is driven by an imbalance of pro-inflammatory effector T cells and Treg cells. COYA-201 was developed using the immunosuppressive cell exosome (iscEXO) platform, derived from Treg cells and M2 macrophages, both of which are prominent anti-inflammatory and neuroprotective cells211.
Exosomes are membrane-derived small vesicles, rich in proteins and RNAs and which can avoid potential cell-based issues such as immune rejection212 and polarization to a pro-inflammatory cell type213. Compared to MSC-derived exosomes, Treg cell-derived exosomes have much higher suppressive capacity and anti-inflammatory function213. The first-in-human trial of COYA-201 was expected in 2022.
Strategies to improve ALS R&D
Clinical trials in ALS have historically seen high failure rates for various reasons (Box 3). We highlight below mitigation strategies for these factors which may lead to significant enhancement of successful therapy development.
Box 3 Reasons for high failure rate in ALS clinical trials Amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA) are the only neurodegenerative diseases for which multiple disease-modifying therapies have been approved, but modified strategies are required to build on these successful initial steps. In the past clinical trials have tended to include patients with ALS as though this is a homogeneous, rather than a heterogeneous disease. Future trials need to subclassify patients with ALS by the upstream driving pathogenetic mechanism (for example, the presence of a specific genetic cause) and by disease progression rate which can be measured by changes per month in the ALS revised functional rating scale–revised score or by newly developed tools227,233. Until now it has not been possible to properly deploy biomarkers of target engagement and therapeutic efficacy and there is a need to advance biomarkers that are emerging from preclinical models into clinical trials. In some recent genetic therapy trials (for example, for SOD1-associated and C9orf72-associated ALS), measurement of specific proteins in cerebrospinal fluid (CSF) (SOD1 and pathological dipeptide repeat proteins such as poly-GP) has been encouraging187,285. In addition, evidence is emerging that levels of neurofilament proteins in both CSF and plasma, which reflect axonal injury, may represent valuable biomarkers of therapeutic efficacy187,286,287,288,289,290. Until recently there has been over-reliance on the SOD1G93A-transgenic mouse model of ALS58. This is a very useful model which replicates many of the clinical and pathological features of ALS. However, only 2% of patients with ALS have a SOD1 mutation, and unlike the disease in most patients with ALS, SOD1-mediated disease is not a TDP-43 proteinopathy8,9,12. The field is now learning to use the SOD1-transgenic mouse more appropriately, but an ongoing challenge is that murine models of other genetic subtypes of ALS have current limitations for therapeutic testing249,252,256. The field is also now recognizing that more careful attention needs to be directed to the whole translational pathway from preclinical study design to early demonstration of target engagement in patients235. An analysis of failed ALS clinical trials also highlighted issues related to pharmacology and trial design291. As an illustration, of 18 large multicentre randomized controlled trials performed between 2004 and 2014 testing ALS disease-modifying drugs, 11 did not consider pharmacokinetics and only one incorporated a marker of target engagement291. In a trial of the selective COXII inhibitor celecoxib, the CSF was analysed for reduction in prostaglandin E2 levels, 2 months after initiation of dosing292. No reduction in prostaglandin E2 levels was observed and indeed the CNS exposure of celecoxib was minimal292. Likewise, of broader neurodegenerative disease trials, only 54% of trials performed between 2010 and 2020 used target engagement biomarkers240. More recent clinical studies have incorporated biomarkers of target engagement, but the ability to confirm that the therapeutic hypothesis has been fully tested in a clinical setting is key to focusing resources in fruitful areas in the future. Clinical studies in ALS have well-defined end points such as ALSFRS-R score and survival, but the lack of a robust surrogate marker of disease progression is a significant issue for the field. Such markers significantly enhanced the pace of drug discovery in multiple sclerosis where gadolinium-enhancing neuroinflammatory lesions were able to robustly predict clinical outcomes. In this regard, a significant effort is underway to validate biomarkers such as NFL, but the full adoption of such markers by regulatory authorities is not yet in place. A further problem in ALS trial design is the high variability in the speed of disease progression, making the design of well-powered studies challenging. As a result, target effect sizes in many historical trials (>40%) may be unrealistic291. Finally, the time to diagnosis in ALS can mean that the disease process has progressed to a stage where preserving motor neuron health may be particularly challenging and strategies to allow earlier intervention with potential neuroprotective agents would be valuable. Late diagnosis is also a challenge for clinical trials. Preclinical in vivo studies and some human trials have shown that early intervention with neuroprotective agents is more effective. The emergence of promising biomarkers such as NFL, as well as effective disease-modifying treatments may encourage earlier referral to specialist ALS clinics.
Improving preclinical studies
Target identification
Major recent advances have taken place that underpin a strong pipeline of candidates for neuroprotective therapy development. Our understanding of disease pathophysiology has increased, with greater knowledge of the genetic architecture of ALS and of the multiple overlapping mechanisms that contribute to motor neuron injury in the presence of specific genetic mutations.
This wealth of additional genetic and pathophysiological information means that there is no shortage of potential novel therapeutic targets. To date successful neuroprotective approaches for ALS are exclusively CNS-penetrant small molecules, which presents challenges for identifying druggable targets. Approaches that draw on the plethora of existing publicly available biological data could help to rapidly identify small molecule targets, a recent example being TargetDB214. Machine learning-based approaches could also be deployed to assist with target identification; for example, the use of knowledge graphs which link disparate types of data with relational inferences derived from natural language processing215. Such strategies have been used to identify COVID-19 therapies216 and are being deployed in ALS to identify new targets, validated using patient-derived cell models (BenevolentAI).
Additional therapeutic modalities are expanding the list of tractable targets, and the successful use of gene therapy (onasemnogene abeparvovec) and ASO (nusinersen) approaches for amelioration of motor neuron injury in spinal muscular atrophy provide realistic prospects for therapeutic advances in defined genetic subtypes of ALS. The limited options for large-molecule (antibody) therapeutics in neurology may be abrogated by new technologies that enable CNS transport of antibody therapeutic agents. This was recently demonstrated in mice and primates using a transferrin-targeting moiety engineered into the Fc portion of a BACE antibody217. This strategy is being applied in several neurodegenerative conditions including FTD (DNL53, Denali Therapeutics; NCT05262023).
Multiple mechanisms, as described above, can contribute to the ultimate demise of motor neurons. This raises the prospect that multiple targets may need to be addressed simultaneously to achieve significant slowing of disease progression. This can be achieved with polypharmacology (for example, AMX0035) and indeed the combination of riluzole and edaravone has become the standard of care for ALS in some countries. A second approach may be to identify single upstream targets that can in turn address multiple downstream mechanisms (for example, M102 which targets the transcription factors Nrf2 and HSF1).
Drug repurposing is often proposed as a potential shortcut to identifying novel therapies and has shown some success for neurological disorders, including dimethyl fumarate for multiple sclerosis218. However, greater consideration of the drawbacks and potential limitations of repurposing is needed218. The most fruitful approach may well be ‘on-target’ repurposing where the original target of the therapy is the intended target for the new indication. However, the need for CNS penetration and whether the risk–benefit and dosing level is appropriate in the new indication are often limiting issues.
Target validation
The use of patient-derived cell models has added an extra dimension to enable early validation of therapeutic hypotheses and complements traditional preclinical in vitro and in vivo models. Fibroblasts donated by patients with ALS can now be reprogrammed into the CNS cells relevant for ALS (for example, motor neurons, astrocytes and microglia). iPSC-derived neurons and glia, where cells are reverted to an embryonic stage of development, have the potential to show early pathophysiological abnormalities. However, direct reprogramming via induced neural progenitor cells has the advantages of generating non-clonal cell lines which retain the features of ageing, of crucial importance in the study of age-related neurodegenerative disorders124,219. These human CNS-relevant cell models are being used to define new therapeutic targets and for the screening of libraries to identify novel neuroprotective therapies220.
Non-mammalian model systems are also emerging as powerful tools to enable target validation. Drosophila melanogaster (fruitfly) and Danio rerio (zebrafish) are increasingly used as models to probe disease mechanisms and have the potential to expand the number of available genetic models and to rapidly validate mechanisms in ALS using genetic or pharmacological approaches221. For example, drosophila models carrying mutations in a range of ALS-related genes have demonstrated a role for dysregulation of RNA metabolism which has been subsequently validated in human biosamples222. Zebrafish carrying C9orf72hexanucleotide repeats were shown to replicate key features of ALS/FTD223 and to complement mammalian models.
Preclinical screening cascades
It is now possible to develop preclinical screening cascades which take us far beyond unreliable readouts of survival in SOD1-transgenic mice. A combined approached can be used, with target-specific screening in vitro validated early in orthogonal patient-derived cell assays for selected compounds. SOD1-transgenic rodents can be used for early mechanistic pharmacokinetic–pharmacodynamic work, followed by studies focused on evaluating more global effects on motor system degeneration. Finally, validation in alternative model systems such as patient-derived in vitro models and more physiologically relevant mouse models should enhance confidence in clinical translatability. Such an approach should enable a step-change in our ability to predict therapeutic efficacy in the clinic (Fig. 3).
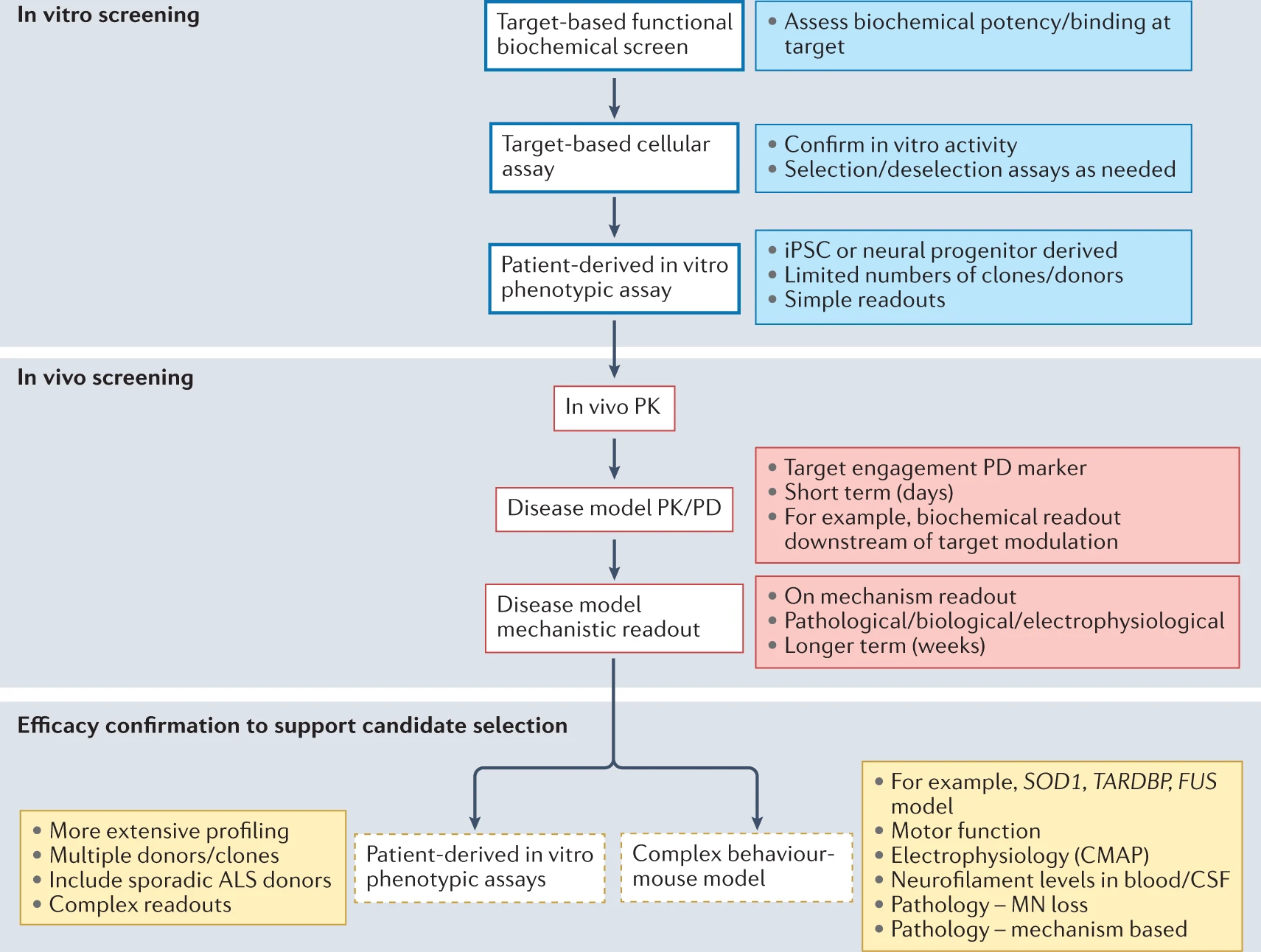
A rational and enhanced preclinical efficacy screening cascade for ALS.
In terms of efficacy assessment and increasing confidence in translation from the preclinical to the clinical arena, an enhanced approach would take advantage of the latest developments in preclinical disease modelling in rodents and non-rodent in vivo systems, as well as patient-derived cellular models. A standard in vitro cascade is shown for a target-based approach, which is subsequently confirmed at an early stage in a relevant patient-derived phenotypic model. Such models can be used as orthogonal screens through a development programme to ensure that efficacy against a relevant disease phenotype is maintained. At later stages in vivo mammalian models can be used as purely mechanistic pharmacokinetic/pharmacodynamic systems to ensure that therapeutic approaches have the required drug metabolism and pharmacokinetics, potency and specificity in vivo. Subsequently, confidence in clinical translation can be obtained in either more extensive screening in patient-derived model systems or complex disease models. Ideally several approaches should be pursued. These complex models with behavioural readouts of motor function would incorporate ‘translational’ biomarkers that have the potential to predict likely clinical benefit, such as measurements of compound muscle action potential (CMAP) and neurofilament light. ALS, amyotrophic lateral sclerosis; CSF, cerebrospinal fluid; iPSC, induced pluripotent stem cell; MN, motor neuron; PD, pharmacodynamic; PK, pharmacokinetic.
Advances in ALS clinical trials
A consensus has emerged that optimizing the use of resources, widening eligibility criteria for trials and minimizing exposure of participants facing a life-threatening disease to ineffective treatments and to placebo are of crucial importance. There has been a recent concerted effort by the international ALS community to address the limitations now recognized in traditional ALS clinical trials methodology. The revised Airlie House consensus guidelines were developed in 2019 to accelerate translational impact for ALS and addressed nine areas of need within ALS research ranging from preclinical research through to trial design and statistical methods, as well as advocating the incorporation of biomarkers and the development of home-based outcome measures for clinical trials224,225. There have been recent promising innovations in trial design, patient selection and randomization, and biomarker development.
Patient stratification
Patient stratification can be based on clinical parameters, genetics, disease stage and the rate of disease progression. Advances in the understanding of the genetic landscape of ALS and overlapping disease pathophysiological mechanisms have accelerated in recent years, as outlined above. Recent exciting progress in precision medicine, exemplified by genetically targeted therapies, is poised to change the natural history of ALS for some subgroups of patients. It is apparent that a precision medicine approach is required to optimize the rapid development of effective neuroprotective therapies for patients with ALS.
A case is emerging for routine genetic subclassification of ALS, regardless of whether a family history is present35,226. We now have improved systems for stratifying patients by disease progression rate224,227 ensuring that patients with fast and slow disease progression can be evenly balanced in trial groups, and simple disease-staging systems228,229 may also add value in future clinical trials. Using data from across 14 European cohorts, an eight-factor validated multivariate model was developed for predicting survival in individual patients with ALS230,231. The use of such a model is predicted to allow up to 80% of patients with ALS to take part in clinical trials, while minimizing the impact of variable rates of disease progression. Thus, prognostic heterogeneity in ALS can now be quantified232. Risk profiles can be used to improve randomization, explore risk-based outcomes and increase statistical power as a covariate in the final analysis of trial data. Stratifying predicted survival into tertiles may be the optimal approach233. In addition, stratifying patients with ALS based on baseline NFL levels is emerging as a useful approach which was recently deployed in the phase III tofersen trial189.
Innovations in trial design and outcome measures
The traditional approach has been the randomized controlled trial in which the effects of the drug candidate are compared to that of placebo, and with survival or ALSFRS-R score as the commonly used primary outcome measures. This approach is costly, time-consuming and inefficient. Many preclinical and early phase I/II trials in ALS have yielded promising results that were not replicated in phase III trials. Substantial consideration has recently been devoted to the refinement of clinical trials in ALS. The ALSFRS-R has been used extensively as an outcome measure, but violates Rasch model expectations and is limited by the multidimensionality that arises from the summation of multiple subscales, which prevents direct comparison of patients with identical total scores233. A new self-reported ALS disability scale has been developed, with improved responsiveness. It is a 28-item Rasch-built overall ALS Disability Scale, with each item scored as 0, 1 or 2, and which demonstrates excellent test–retest reliability227. Other novel outcome measures are in development including modifications to the ALSFRS-R.
Multi-arm, multistage platform trials (MAMS) that incorporate biomarkers of target engagement/therapeutic efficacy are now poised to accelerate drug discovery and increase trial participation. In a MAMS trial with a sequential adaptive design, the sample size is not fixed in advance and the emerging trial data are sequentially analysed with in-built, pre-determined futility and superiority analyses. This enables ineffective treatment arms to be discontinued and studies with a positive signal to seamlessly graduate to the next trial phase. Platform trial designs (for example, Healey ALS Platform Trial and MND-SMART) have emerged to allow more rapid assessments and inclusivity for patients, and more efficient deployment of a pooled placebo arm233,234.
A further European platform trial, TRICALS, will start imminently as several of the initial portfolio of trials are recruiting patients in multiple European centres.
A master protocol allowing the simultaneous evaluation of multiple compounds and efficient use of a common placebo group has been successfully used in other disease areas, most notably oncology235. However, it is noteworthy that in these conditions robust biomarkers of disease burden are available. Such biomarkers have not yet been established in ALS, although promising candidates are now emerging; for example, neurofilaments and specific protein biomarkers in genetic therapy trials.
These recent proposals to modify the traditional design of ALS clinical trials have the potential to reduce the sample size required and the placebo exposure time for trial participants, as well as the cost, burden for patients and the trial duration232. Event-driven trial designs may ameliorate the consequences of inaccurate baseline assumptions for trial design and the resulting unknown trial duration can be mitigated by deployment of planned interim analyses.
Improved patient-reported outcome measures, including home assessments, are predicted to improve the reliability and sensitivity of trial end points. Evidence is emerging that home assessments may be a viable strategy for measuring the disease trajectory in ALS clinical trials232,236,237. This strategy has been accelerated by the shift towards telemedicine for the provision of clinical care to patients with ALS during the COVID-19 pandemic.
Dialogue with regulatory bodies will be essential in bringing to fruition these proposed innovations for ALS clinical trials. It is noteworthy that the FDA has already published guidance for adaptive platform master protocol trial designs238 and the EMA has established a plan to carefully explore this issue.
Biomarkers
Robust biomarkers are needed in ALS to measure disease progression and provide prognostic information, to identify potential therapeutic targets, to stratify patients prior to clinical trial entry, and as measures of target engagement and therapeutic efficacy in clinical trials. A large volume of biomarker research has been undertaken, but only a few robust, validated biomarkers have so far emerged (Box 4). Recent reviews have comprehensively described the current state-of-the-art for clinical, biochemical, imaging and electrophysiological biomarkers225,239.
Box 4 ALS biomarkers There has been important recent progress in the identification of useful biochemical biomarkers for amyotrophic lateral sclerosis (ALS). In SOD1-associated ALS, gene silencing approaches have been shown to decrease the levels of the SOD1 protein in the central nervous system (CNS) and cerebrospinal fluid (CSF) in mouse models186. In a recently reported phase II trial of tofersen, an antisense oligonucleotide (ASO) intrathecally administered therapy that aims to lower the level of SOD1, there was a dose-dependent reduction of the SOD1 protein in the CSF by a mean of 33% in patients receiving the highest dose of 100 mg. Trials of tofersen have also shown the potential value of neurofilament light (NFL) levels in blood and CSF as a biomarker of motor neuron injury and therapeutic efficacy187,189. In relation to C9orf72-ALS, the dipeptide repeat protein poly-GP may emerge as an important pharmacodynamic biomarker. The levels of this protein in the CSF seem to be relatively stable in patients with C9orf72-ALS over time, and decreased levels have been observed following ASO therapy in cellular and mouse models of C9orf72-ALS257. Thus, in genetic therapy trials, measurement of the protein encoded by the gene being targeted is proving to be a valuable biomarker in both preclinical studies and human trials. However, in the terminated phase I trial of a C9orf72-ASO sponsored by Biogen (NCT03626012), robust dose-dependent lowering of dipeptide repeat proteins was achieved, but with no evidence of benefit in terms of clinical end points and neurofilament levels191. The most promising biochemical biomarkers identified to date are neurofilament proteins, which are neuronal cytoskeletal components released into the CSF and blood as a consequence of axonal injury, irrespective of the primary causative event. Recent data have indicated that measurement of neurofilament protein levels, particularly NFL, has great potential as a robust biomarker in ALS. Measurement of NFL has been shown to have value in the diagnosis of ALS, in prediction of the rapidity of the disease course and in pharmacodynamic monitoring187,293,294. Levels of NFL in CSF and plasma are tightly correlated, which allows serial sampling to be undertaken less invasively from blood. A recent large, multicentre, clinic-based, longitudinal cohort study measured NFL levels serially in 258 patients with ALS and in disease controls and healthy individuals. The first-visit NFL level was strongly associated with the rate of disease progression, independently of other prognostic factors. Modelling of plasma NFL as an outcome measure in clinical trials highlighted that, in comparison with the use of the ALSFRS-R, smaller sample sizes would be required and earlier detection of disease slowing would be facilitated295. It is possible that neurofilament levels may enable the identification of a subset of responsive patients when a new neuroprotective agent is being tested in a trial population with variable upstream pathophysiological causes of motor neuron injury, as in sporadic ALS. NFL levels may also help stratify trial patients into more homogeneous groups. In addition, NFL has been shown to be a biomarker of pre-symptomatic ALS and phenoconversion, in carriers of pathogenic SOD1 and C9orf72 mutations, and this will aid in the design and conduct of early intervention and prevention trials296. The value of undertaking transcriptomics analysis of peripheral blood mononuclear cells to detect gene expression biomarkers of good and poor responsiveness to an experimental compound, as well as a panel predicting the level of response, has recently been shown in a phase II trial of low-dose IL-2 as an anti-inflammatory agent for the treatment of ALS198. MicroRNAs have promise as biomarkers, although there is a lack of consistency in the specific microRNAs identified as being differentially expressed in ALS across different laboratories, and few longitudinal studies have been conducted. A recent study found that high levels of miR-181 in plasma predicted an increased risk of death in an independent discovery patient cohort and a replication patient cohort. Combining the measurement of miR-181 with NFL gave enhanced prognostic accuracy297. Electrophysiological biomarker indices have also shown some promise. Motor axonal degeneration (for example, decrease in compound muscle action potential) can be found in patients and animal models of ALS252. The sensitivity and reliability of the motor neuron number index (MUNIX) were investigated in a longitudinal multicentre study298. The neurophysiological index has been proposed as a quantitative measure of peripheral disease burden in ALS and has been found to decline faster than other commonly used measures of disease progression, with sensitivity to change in as little as 4 weeks. The neurophysiological index has been highlighted as having the potential to expedite completion of phase II ALS trials299,300. Imaging studies have to date tended to show group differences rather than changes at the level of individual patients with ALS. Validated imaging biomarkers sensitive to and specific for disease change at the individual level have remained elusive. Imaging biomarkers that have potential usefulness in future trials include PET ligands and magnetic resonance spectroscopy. Proton magnetic resonance spectroscopy (MRS) can provide insights into the biology of the CNS through measurement of metabolites (for example, the neuronal marker N-acetyl aspartate (NAA)), the glial marker myoinositol, choline-containing compounds, amino acids and neurotransmitters (including glutamate and GABA), and measurement of bioenergetic status (such as derivatives of ATP) and glutathione (as a measure of oxidative status)239. Brain MRS studies have shown a reduction in NAA correlating with the burden of upper motor neuron pathology301, and changes in bioenergetic status have been found in the CNS and muscle of patients with ALS89. However, studies measuring glutamate, myoinositol, choline, GABA and glutathione have so far produced inconsistent results239, and it is clear that rigorous standardization of MRS acquisition methodology is required to take this field forward. PET, either alone or combined with MRI (PET–MRI), also holds some promise for disease monitoring in clinical trials. 18F-Fluoro-2-deoxy-2-D-glucose (FDG) PET measures cellular glucose uptake, and has been used to assess cerebral metabolic activity, with decreases documented in the motor, premotor and prefrontal cortices and the basal ganglia in patients with ALS302. A recent FDG PET study showed glucose metabolic changes in pre-symptomatic carriers of C9orf72 mutations at the individual level, which preceded the onset of symptoms and elevations in neurofilament levels303. The 18-kDa TSPO/peripheral benzodiazepine receptor is thought to be expressed specifically by activated microglia and astrocytes. Enhanced microglial activation has been found in multiple areas of the brain in patients with ALS304 and represents a potential way to monitor interventions targeting neuroinflammation. Recently developed novel radioligands show promise for in vivo PET imaging of the brain; for example, measurement of oxidative stress arising from mitochondrial dysfunction using 62Cu-ATSM305 and quantification of synaptic density using 11C-UCB-J306.
Outlook
Significant recent advances have been made in understanding the genetic architecture and pathophysiology of ALS. A strategy has emerged where more accurate subclassification of patients can be made based on identification of risk genes beyond the traditional boundaries of sALS and fALS. This expanded genetic understanding has also enabled the validation of several pathophysiological biological pathways for therapeutic targeting and, alongside the expansion of therapeutic modalities, there is now no shortage of therapeutic hypotheses to explore preclinically.
In the preclinical space, the number, variety and clinical relevance of available model systems to enable more rigorous preclinical screening have expanded. This enables validation of therapeutic hypotheses and targets across models and reduces the risk of failure due to lack of efficacy.
In the clinical space, recent innovations in trial design will enhance outcome measures, patient selection and randomization, minimize the impact of clinical variability and increase statistical power. Platform trials and patient-reported outcomes have the potential to improve the pace of validation of therapeutic agents. This is particularly the case if combined with surrogate biomarkers of disease burden, which are now emerging.
The development of biomarkers of target engagement and efficacy to bridge the gap from preclinical to clinical testing is a key future need, as is the application of sound pharmacological principles in therapy development. Several biomarkers (biochemical, physiological, imaging) that can be applied across the translational pathway show great promise.
Of added importance is the continued impact of patient advocacy groups encouraging and financially supporting researchers and clinicians. This strong and visible advocacy has been a factor in elevating the priority of regulatory agencies to address the high unmet need of individuals facing ALS. For example, regulatory agencies appear to be more receptive to considering adequate and well-controlled phase II data as part of a streamlined approval process, as was recently shown with AMX0035. Having the attention and guidance of regulatory agencies continues to be an important factor in promoting rapid access to new therapies for patients with ALS.
In summary, there is great potential for developing improved neuroprotective treatments for ALS in the near future. We advocate for academia–industry partnerships which accelerate the pace of rigorous testing of therapeutic hypotheses, incorporating the latest advances in preclinical screening, biomarker discovery and trial design. The unmet need of patients with ALS is clear and a range of tools are now poised for effective translation.