
Amyloid-associated increases in soluble tau relate to tau aggregation rates and cognitive decline in early Alzheimer’s disease

By Alexa Pichet Binette, Nicolai Franzmeier, Nicola Spotorno, Michael Ewers, Matthias Brendel, Davina Biel, Olof Strandberg, Shorena Janelidze, Sebastian Palmqvist, Niklas Mattsson-Carlgren, Ruben Smith, Erik Stomrud, Rik Ossenkoppele, and Oskar Hansson
Excerpt from the article published in Nature Communications 13, 6635, 04 November 2022. DOI: https://doi.org/10.1038/s41467-022-34129-4
Editor’s Highlights
- Amyloid-β (Aβ) plaques start to accumulate ~20 years before symptom onset followed by the accumulation and spreading of neurofibrillary tau aggregates with ensuing neurodegeneration and AD dementia.
- Initially, the Aβ-related increase of p-tau seeds is taken up by neurons to initiate the misfolding and aggregation of tau in a given brain region, with more substrate (i.e. soluble p-tau) for aggregation leading to more rapid local tau aggregation.
- Later on, local self-replication and accumulation of misfolded tau aggregates may take over once a critical threshold of tau aggregates is reached.
- Targeting Aβ fibrils and soluble p-tau in early AD may be a promising strategy to slow the formation of tau aggregates, and thereby preserve cognitive abilities for a longer period.
- Anti-Aβ therapies promptly reduce soluble p-tau concentrations in both CSF and plasma, reinforcing the close relationship between Aβ pathology and increases in extracellular p-tau levels
Abstract
For optimal design of anti-amyloid-β (Aβ) and anti-tau clinical trials, we need to better understand the pathophysiological cascade of Aβ- and tau-related processes. Therefore, we set out to investigate how Aβ and soluble phosphorylated tau (p-tau) relate to the accumulation of tau aggregates assessed with PET and subsequent cognitive decline across the Alzheimer’s disease (AD) continuum. Using human cross-sectional and longitudinal neuroimaging and cognitive assessment data, we show that in early stages of AD, increased concentration of soluble CSF p-tau is strongly associated with accumulation of insoluble tau aggregates across the brain, and CSF p-tau levels mediate the effect of Aβ on tau aggregation. Further, higher soluble p-tau concentrations are mainly related to faster accumulation of tau aggregates in the regions with strong functional connectivity to individual tau epicenters. In this early stage, higher soluble p-tau concentrations is associated with cognitive decline, which is mediated by faster increase of tau aggregates. In contrast, in AD dementia, when Aβ fibrils and soluble p-tau levels have plateaued, cognitive decline is related to the accumulation rate of insoluble tau aggregates. Our data suggest that therapeutic approaches reducing soluble p-tau levels might be most favorable in early AD, before widespread insoluble tau aggregates.
Introduction
Alzheimer’s disease (AD) is characterized by a cascade of molecular and neurodegenerative brain changes, in which Aβ plaques start to accumulate ~20 years before symptom onset followed by the accumulation and spreading of neurofibrillary tau aggregates with ensuing neurodegeneration and AD dementia1,2. Multiple neuroimaging studies using positron emission tomography (PET) to track in vivo pathological processes in AD showed that cortical Aβ deposition precedes neocortical tau aggregation by several years1,3, and Aβ has been shown to amplify tau spreading in preclinical studies4,5. Among the earliest tau-related abnormalities in AD are increases in soluble hyperphosphorylated tau (p-tau) concentrations, which can reliably be detected in cerebrospinal fluid (CSF)1 and precede the neocortical deposition of insoluble fibrillary tau pathology in AD6,7,8. The rate of cellular tau production9and soluble p-tau increases10 have been shown to correlate with Aβ burden, further strengthening the view that Aβ systematically induces p-tau increases. Recent studies could show that levels of p-tau start increasing already at the preclinical stage of AD when persons are asymptomatic11, increase even further in early symptomatic AD and reach a plateau in patients with AD dementia12,13. Importantly, levels of soluble p-tau have been shown to correlate with neuropathological levels of insoluble fibrillar tau14,15,16, and preclinical studies found that soluble hyperphosphorylated p-tau seeds can induce a cascade of tau aggregation in animal models4,17,18,19. Therefore, increases in soluble p-tau may drive subsequent tau aggregation and spread in AD, suggesting p-tau as a potential treatment target to halt tau spread and the formation of toxic insoluble tau aggregates. However, the cross-link between Aβ, p-tau and tau aggregation is yet to be better understood in humans across the spectrum of AD.
A putative mechanism by which Aβ triggers tau secretion is via stimulation of neuronal activity20. Extracellular Aβ has been consistently shown to induce neuronal hyperactivity21,22and neuronal activity enhances tau secretion17,23,24. Therefore, a major pathway for the propagation of tau pathology in AD are synaptic connections, which are assumed to provide the roadmap for trans-neuronal tau spreading25,26. This is supported by preclinical work, showing that injected p-tau seeds can be taken-up by post-synaptic neurons and spread across anatomically connected rather than spatially adjacent brain regions4,17,18,19, further supporting soluble p-tau as a key driver of tau propagation. We recently translated these preclinical findings to in vivo neuroimaging data by combining functional MRI-based connectomics with tau-PET in AD. We and others reported that patient-level tau accumulation is related to the connectivity patterns of regions where tau aggregates emerge first (i.e, tau epicenters)27,28,29.
Altogether, the literature supports a view in which i) Aβ is linked with increased concentrations of soluble p-tau early in the AD continuum, ii) greater concentrations of soluble p-tau are associated with insoluble tau aggregates, and iii) tau aggregates expand across connected brain regions. Yet, evidence in human studies overwhelmingly stem from cross-sectional studies, and a biologically plausible cascade model that incorporates these individual findings and can link soluble p-tau with downstream tau aggregation and clinical manifestations across the entire AD spectrum is still missing. Testing this model is critical to clarify the potential of p-tau as a target to attenuate tau aggregation and spread, and the optimal target populations that may benefit from such treatments. This is particularly important since drugs efficiently lowering the soluble tau levels have recently advanced to phase 2 (Clinical Trial ID: NCT05399888). Based on progress in the anti-tau drug development pipeline, it is essential to establish during which phases of the disease soluble p-tau levels are most associated with subsequent accumulation of insoluble tau aggregates and cognitive decline.
To address these knowledge gaps, we leveraged both cross-sectional Aβ and CSF p-tau levels, resting-state fMRI connectomics, longitudinal tau-PET (measuring accumulation of insoluble tau aggregates) and longitudinal cognitive assessments (measuring cognitive decline) in two independent large-scale AD cohort studies. We first tested how soluble p-tau concentrations measured in CSF and local Aβ fibrils at baseline affected the local accumulation rate of tau aggregates over time across the brain. Second, adding resting-state functional connectivity, we investigated if soluble p-tau was related to connectivity-based accumulation of tau aggregates across the brain. Lastly, we investigated those biomarker changes leading to cognitive decline, bringing together both measures of soluble p-tau and tau aggregates accumulation as they follow more closely the apparition of symptoms than Aβ pathology30,31. Informed by the results we propose a working model of pathophysiological AD progression that is largely built on longitudinal in vivo data, from the early preclinical phase towards the clinical syndrome of AD dementia. Overall, our study suggests that Aβ-related increased soluble levels of p-tau is a key driver in the accumulation of tau aggregates and connectivity-mediated tau spreading in early-stage preclinical and prodromal AD, while the accumulation rate of tau aggregates becomes self-promoting in late-stage AD dementia and might no longer be driven by p-tau. We thus suggest that soluble p-tau levels may be an optimal treatment target for attenuating tau aggregation and subsequent cognitive decline in the earliest stages of AD, prior to the development of widespread insoluble fibrillar tau aggregates and dementia, which will be critical for the design of future clinical trials.
Results
Study design and participants
The study was conducted in one of the largest samples available worldwide with cross-sectional Aβ-PET and soluble CSF p-tau measurement, and longitudinal tau-PET and cognition, covering the full spectrum of AD. The Swedish BioFINDER-2 cohort was the primary cohort of interest, from which participants with all abovementioned measures were included. The final sample included 400 participants composed of 204 cognitively unimpaired (CU) Aβ-negative participants, 65 Aβ-positive CU, 65 Aβ-positive patients with mild cognitive impairment (MCI) and 66 Aβ-positive individuals with AD dementia (Table 1). The CU and MCI Aβ-positive participants were grouped together to study the early stages of AD, hereafter referred to as “non-demented participants”. All main analyses were also conducted in each group separately, with all results in Supplementary information. To quantify the regional levels of insoluble tau aggregates, tau-PET was performed using the second-generation radiotracer [18F]RO948, shown to be sensitive to early tau aggregates32,33. Insoluble Aβ aggregates were measured with Aβ-PET using [18F]flutemetamol as the radiotracer. All PET data were parcellated in 200 cortical regions-of-interest (ROI) using the Schaefer brain atlas34. For analyses done across the 200 brain regions, only results surviving multiple comparisons from false discovery rate are reported. Concentrations of soluble p-tau were measured using p-tau217 in the CSF35. Accumulation of tau aggregates was quantified as the rate of change in tau-PET retention over time (SUVR/year) in each brain region separately, derived from linear mixed effect models. Cognitive decline was quantified as rate of change on cognitive scores (a cognitive composite score analogous to the PACC5 and MMSE) per year derived from linear mixed effect models. The main results focusing on early AD were also validated in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database using [18F]flortaucipir PET, CSF p-tau181 as well as [18F]florbetapir or [18F]florbetaben Aβ-PET (n = 119: 52 CU Aβ-negative, 36 Aβ-positive CU and 31 Aβ-positive MCI participants; all figures and extended results in Supplementary information). Given the smaller sample size in ADNI, only analyses where results could be detected with sufficient power calculated based on the BioFINDER-2 analyses were conducted.
Aβ-negative controls (n = 204) | Aβ-positive non-demented (n = 130) | Aβ-positive AD dementia (n = 66) | |
|---|---|---|---|
| Age (years) | 62.4 ± 15.0 | 71.7 ± 8.4 | 72.7 ± 7.3 |
| Sex F (%F) | 100 (49%) | 63 (49%) | 37 (56%) |
| Education (years) | 12.7 ± 3.2 | 12.4 ± 4.4 | 11.8 ± 4.3 |
| APOEε4 carriers (%) | 73 (36%) | 95 (73%) | 44 (67%) |
| CSF p-tau217 (pg/ml) | 47.6 ± 31.4 | 243.7 ± 174.5 | 570.8 ± 300.9 |
| MMSE | 29.0 ± 1.2 | 27.7 ± 1.9 | 20.1 ± 4.4 |
| Cognitive composite score | 0.01 ± 0.7 | −1.4 ± 1.2 | −4.4 ± 1.5 |
| tau-PET follow-up time (years) | 2.0 ± 0.6 | 2.0 ± 0.6 | 1.7 ± 0.3 |
| Number of PET scans, n (%) | |||
| 2 | 173 | 92 | 48 |
| 3 | 29 | 32 | 18 |
| 4 | 2 | 6 | 0 |
| Cognitive follow-up time (years) | 2.4 ± 0.8 | 3.1 ± 0.7 | 2.0 ± 0.6 |
| Number of cognitive assessments, n (%) | |||
| 2 | 144 | 13 | 11 |
| 3 | 42 | 52 | 22 |
| 4 | 12 | 46 | 33 |
| 5 | 4 | 19 | 0 |
BioFINDER-2 cohort demographics
Data are presented as mean ± standard deviation unless specified otherwise. One Aβ-positive non-demented participant had missing APOE genotype. Cognitive composite are z-scores. See Supplementary Table 1 for ADNI participants.
Aβ beta-amyloid, APOEε4 apolipoprotein E genotype (carrying at least one ε4 allele), CSF p-tau217 cerebrospinal fluid phosphorylated tau 217, MMSEMini-Mental State Examination, PET positron emission tomography.
n the BioFINDER-2 cohort, the baseline distribution of insoluble tau aggregates assessed via tau-PET recapitulated the AD-typical deposition in the medial and lateral temporal lobes in the controls and non-demented participants, and into lateral and medial parietal and lateral occipital regions at symptomatic AD stages (Fig. 1a and Supplementary Fig. 1 for CU and MCI separately). The longitudinal accumulation rate of tau aggregates (i.e., the annual tau-PET rate of change measured over a mean time of 2 years) was highest in temporal lobe regions in non-demented participants, and showed more extensive involvement mainly of medial parietal and lateral frontal regions in AD dementia (Fig. 1b and Supplementary Fig. 1). Quantitatively, focusing on regions corresponding to a temporal meta-ROI encompassing medial and lateral parts of the temporal lobe – key regions of tau aggregates early in the disease process and approximating Braak stages I to IV -, the average annual SUVR rate of change was 0.7% in Aβ-negative controls, 3.3% in Aβ-positive non-demented individuals, and 9.0% in Aβ-positive patients with AD dementia. Between-group comparisons showing regions where accumulation of tau aggregates differ are also reported in Supplementary Fig. 2 for complementary description of regional differences of tau-PET rate of change.
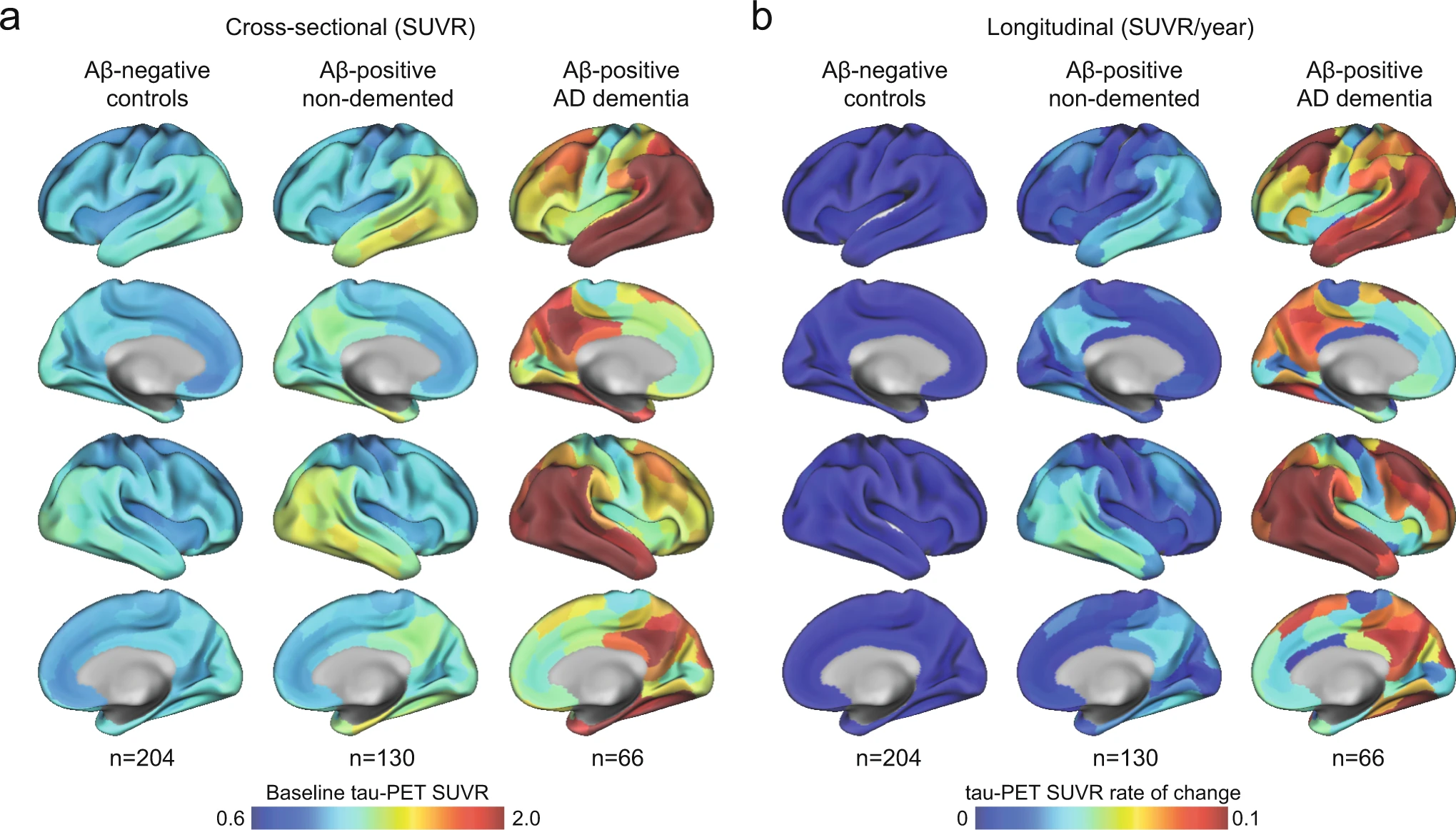
Mean spatial distribution of cross-sectional tau-PET [18F]RO948 SUVR and longitudinal rate of change.
a Surface renderings of average baseline tau-PET SUVR in Aβ-negative controls, Aβ-positive non-demented participants and Aβ-positive patients with AD dementia in the 200 parcels from the Schaefer 200-ROI atlas. b Surface renderings of yearly tau-PET SUVR rate of change derived as the slope from linear mixed-effect models in the same participants group as in a. The group of Aβ-negative controls spans both middle-aged and older individuals. In Supplementary Fig 1, we show individuals above 50 years old, as well as dividing the non-demented group into CU and MCI. Source data are provided as a Source Data file. Aβ beta-amyloid, AD Alzheimer’s disease, CU cognitively unimpaired, MCI mild cognitive impairment, PET positron emission tomography, SUVR standardized uptake value ratio.
In ADNI (Supplementary Table 1), the spatial pattern of the main regions with highest tau-PET binding at baseline and the highest rate of change in tau aggregates per year mirrored the patterns found in BioFINDER-2 (Supplementary Fig. 3). However, the magnitude of accumulation of tau aggregates was almost twice as small compared to BioFINDER-2.
Increased soluble p-tau is the main modifier of tau aggregate accumulation rates in early AD
First, we tested whether either the level of local Aβ aggregates and/or the concentrations of soluble p-tau were most strongly associated with local increases in insoluble tau aggregates over time in early stages of AD using linear regression models in each of the 200 brain regions. Across Aβ-positive non-demented participants, regional Aβ was positively associated with greater accumulation of tau aggregates over time, most prominently in temporo-parietal regions (Fig. 2a). On average across the 82 regions surviving correction for multiple comparisons, the standardized estimate of Aβ was 0.26 (range 0.21 to 0.41, average p-values in those regions = 0.03). Levels of soluble p-tau was also positively correlated with accumulation of tau aggregates, but in contrast to regional Aβ, this was observed in nearly all brain regions (n = 194) and the effect sizes were substantially higher (Fig. 2a, average standardized estimate: 0.47, range 0.19 to 0.71, average p-values <0.001). When using regional Aβ and soluble p-tau simultaneously as predictors (Fig. 2b), the widespread effect of CSF p-tau on the accumulation rate of tau remained virtually the same. In contrast, regional Aβ had almost no independent effect on tau accumulation after accounting for soluble p-tau. Of note, a similar effect of Aβ being no long significant after accounting for soluble p-tau was found on baseline tau aggregates (Supplementary Fig. 4). Furthermore, all key regions where soluble p-tau was most strongly associated with accumulation of tau aggregates over time remained significant even when additionally accounting for baseline levels of tau aggregates in each region (Fig. 2c), albeit with slightly lower estimates (average standardized estimate: 0.21, range: 0.05 to 0.44, average p-values = 0.007). These results were also consistent across different measures of Aβ, i.e. if using global Aβ load assessed by PET or the CSF Aβ42/40 ratio instead of regional Aβ aggregates in regression models (Supplementary Fig. 5). Further, investigating the same relations in the CU and MCI groups separately, the strong effect of soluble p-tau on subsequent tau aggregation rate, above the effect of Aβ, was clearly found in both groups (Supplementary Fig. 6). When further adjusting for local baseline tau aggregates, soluble p-tau also remained a significant predictor in temporo-parietal regions in each group (Supplementary Fig. 6).
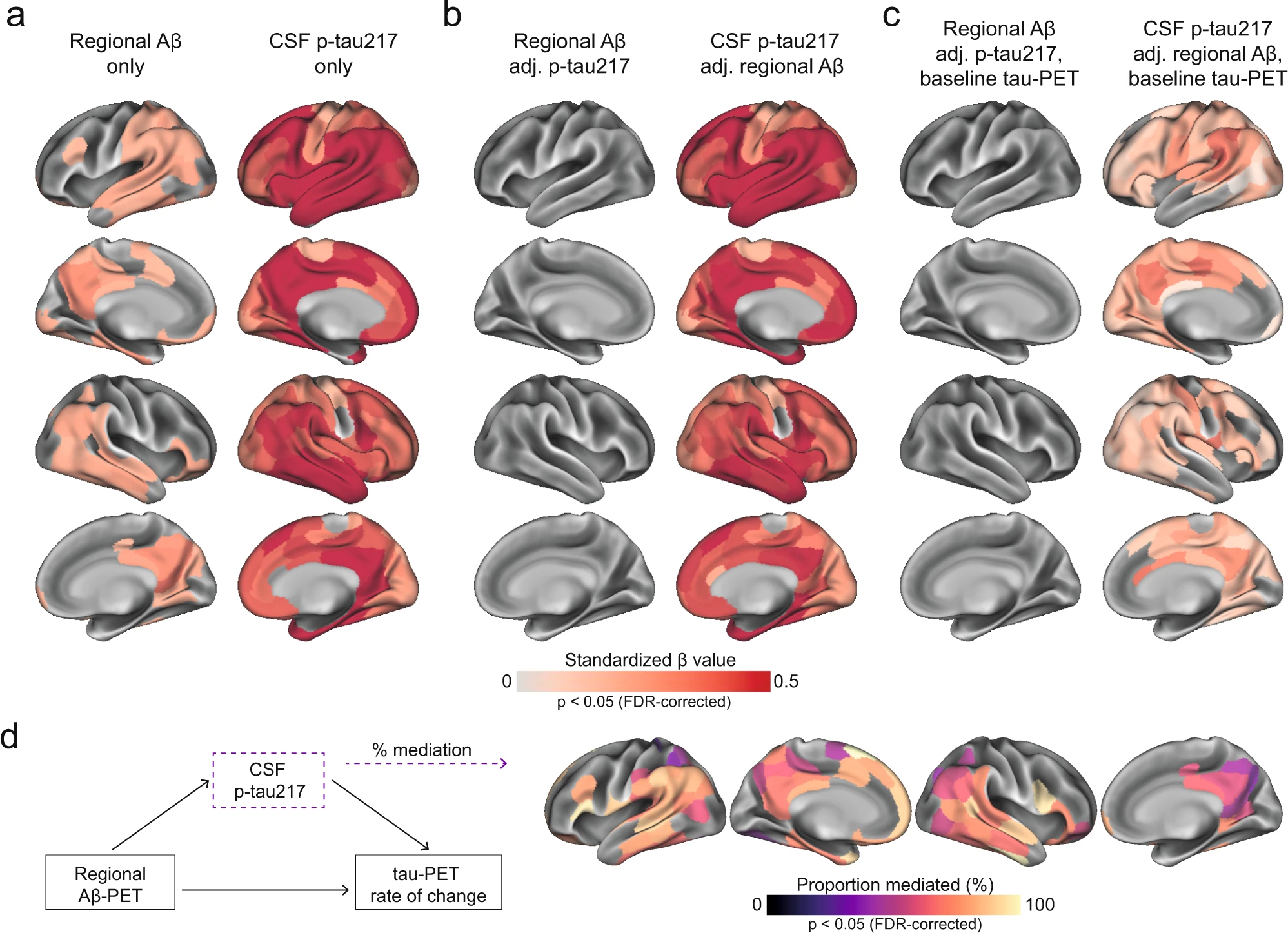
Regional Aβ-PET and CSF p-tau217 associations with regional tau-PET [18F]RO948 rate of change in Aβ-positive non-demented participants.
a Standardized beta coefficient of local Aβ-PET in regions where regional Aβ-PET flutemetamol SUVR (left column) relates to regional tau-PET rate of change, adjusting for age and sex. Right columns were derived from a similar model, but using CSF p-tau217 as predictor instead of Aβ-PET. b Standardized beta coefficient where local Aβ-PET (left column) and CSF p-tau217 (right column) is associated to regional tau-PET rate of change when including both biomarkers in the same model, adjusting for age and sex (tau PET rate of change ∼ regional Aβ-PET + CSF p-tau217 + age + sex). c Same depiction as in b, when additionally controlling for regional baseline tau-PET SUVR. d Mediating effect of CSF p-tau217 on local Aβ-related accumulation of local tau aggregates. The mediation models were performed region-wise, and the percentage of the mediating effect are projected on the brains. Statistical details of the mediation models are shown in Supplementary Fig. 8. Significance of the mediation effect was tested using 1000 bootstrapping iterations. All regions shown on the brain are significant at p < 0.05 after FDR-correction from two-sided statistical tests. Source data are provided as a Source Data file. Aβ beta-amyloid, CSF cerebrospinal fluid, FDR false discovery rate, PET positron emission tomography, p-tau phosphorylated tau, SUVR standardized uptake value ratio.
Lastly, based on the observed interplay between local Aβ and soluble p-tau on the increased rate of tau aggregates accumulation, we formally tested the mediating effect of soluble p-tau on Aβ-related tau-PET rate of change at the regional level. Figure 2d summarizes the proportion of mediating effect of soluble p-tau on all regions surviving multiple comparisons (n = 82), which averaged to 70% across all regions. Overall, the results indicate that the effects of local Aβ aggregates on the accumulation rate of insoluble tau aggregates over time is largely mediated by increased concentrations of soluble p-tau.
In ADNI, as in BioFINDER-2, soluble p-tau, measured with CSF p-tau181, was the main factor related to regional accumulation of tau aggregates over time (Supplementary Fig. 7). Analyses were conducted using two Aβ measures: the global centiloid (CL) score since two different PET Aβ tracers are used in ADNI, and CSF Aβ42. In both cases, soluble p-tau remained significant when accounting for Aβ and baseline tau-PET SUVR in temporo-parietal regions, although the regional pattern was more restricted than in BioFINDER-2 (Supplementary Fig. 7).
Soluble p-tau levels relate to connectivity-based accumulation of tau aggregates in early AD
Another important modifier of regional tau accumulation is the functional architecture of the brain. We therefore investigated whether the association between connectivity-based accumulation of tau aggregates was influenced by the concentration of soluble p-tau. Briefly, after defining participant-specific tau-PET epicenters (i.e., the top 10 regions with the highest tau-PET SUVR probability from Gaussian-mixture modeling at baseline), accumulation of tau aggregates in the remaining 190 regions was correlated to functional connectivity strength to the epicenters27,36. We first applied the model at the group-level, where we found greater accumulation rates of insoluble tau aggregates in regions more strongly connected to the epicenters (shorter distance-based connectivity) in early AD (Fig. 3a). Next, we applied this model at the individual level, defining tau epicenters for each participant. We found that higher accumulation rates of tau aggregates were observed in regions that showed strongest connectivity to the tau epicenters defined on baseline tau-PET, as evidenced by negative β-values (i.e. reflecting the association between connectivity to the epicenters and tau aggregates accumulation across all non-epicenter ROIs; Fig. 3b).
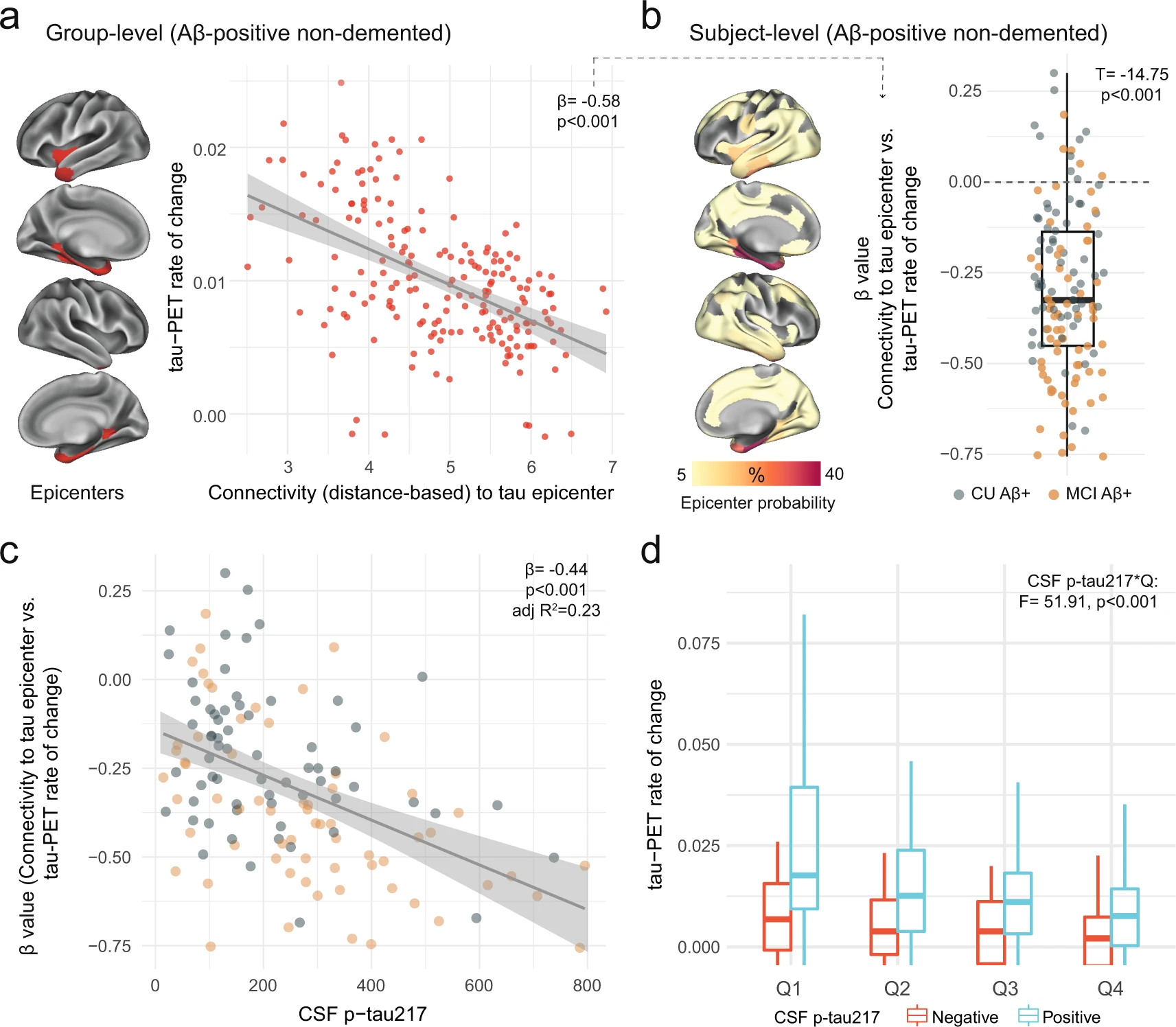
Individualized connectivity-based associations of tau-PET rate of change over time and CSF p-tau217 in Aβ-positive non-demented participants.
a Group-level analysis showing how connectivity to the tau epicenters (projected on the glass brains) relates to tau-PET rate of change across the whole brain. Each dot represents a brain region. Regions more strongly functionally connected to the epicenters have greater rate of tau-PET accumulation. bRepeating the same approach depicted in a at the individual level, the values on the glass brains represent the percentage that each region is classified as an epicenter. The box plot shows the individual β-value (n = 130) from the correlation between tau-PET rate of change and connectivity-based distance to epicenters across all brain regions. c Scatter plot of the associations between CSF p-tau217 and the β-values of epicenter connectivity to tau-PET rate of change. Each dot represents an individual (n = 130). The expected negative association suggests that higher CSF p-tau217 is associated with the overall pattern of tau-PET change in more functionally connected regions to epicenters. d Average tau-PET rate of change across all participants (n = 130) in regions split into quartiles based each region’s connectivity to the tau epicenters (Q1 represents top 25% regions with strongest functional connectivity to the epicenters, etc.). All linear regressions performed were two-sided, without adjustment for multiple comparisons and error bands correspond to the 95% confidence interval. In all box plots, the box limits represent the interquartile range and the line depicts the median value. Aβ beta-amyloid, CSF cerebrospinal fluid, PET positron emission tomography, p-tau phosphorylated tau, Q quartile.
Next, given the association between soluble p-tau and the accumulation of tau aggregates shown in Fig. 2, we hypothesized that higher soluble p-tau concentrations would relate to a stronger association between connectivity to the epicenters and the accumulation rates of tau aggregates across brain regions. In all non-demented Aβ-positive participants, we found that participants with higher soluble p-tau levels had a stronger association between connectivity to epicenter and tau-PET rate of change in non-epicenter ROIs (β-value), while accounting for global Aβ, age and sex (Fig. 3c). This association also survives adjustment for baseline tau-PET (β = −0.23, p = 0.04). The same associations were found in CU and MCI participants alone (Supplementary Fig. 9), suggesting that this effect is not only due to the MCI participants who tend to show greater association between connectivity to epicenter and tau-PET rate of change (more negative β-values on Fig. 3b). Analyzing the data in a complementary way, we measured the rate of tau aggregates accumulation in regions split into quartiles defined at the individual level, i.e. average tau-PET rate of change in top 25% regions with the greatest connectivity to tau epicenters as quartile 1 (Q1), up to quartile 4 (Q4, regions with the lowest connectivity to tau epicenters). Repeated measures ANOVA revealed an interaction of soluble p-tau levels and quartiles (F = 51.9, p < 0.001), suggesting the importance of soluble p-tau and connectivity-based regions on the accumulation rates of insoluble tau aggregates (Fig. 3d). This effect was particularly evident Q1, which showed the greatest effect size between CSF p-tau-positive and p-tau-negative participants (Cohen’s d = 0.70 vs. 0.57 to 0.61 in Q2 to Q4).
Results were also validated in the Aβ-positive non-demented participants from ADNI (Supplementary Fig. 10). The overall connectivity-based association with tau-PET rate of change (β-value) was related to the levels of soluble p-tau (standardized coefficient = −0.24, p = 0.01), there was an interaction between soluble p-tau and connectivity-based tau aggregates accumulation by quartiles (p = 0.01).
Accumulation of insoluble tau aggregates mediates the associations between soluble p-tau and cognitive decline in early AD
With soluble p-tau being related to local accumulation of tau aggregates and connectivity-mediated tau accumulation, we then investigated how those different tau measures related to cognitive decline in early stages of AD (i.e. Aβ-positive non-demented participants). We focused on tau-PET rate of change in regions most strongly connected to the epicenters (i.e. Q1), to capture individualized level of tau aggregates accumulation, and all results were also validated using a commonly used temporal meta-ROI (see Supplementary Fig. 11). All different tau-related measures (CSF p-tau217, tau-PET rate of change in Q1 and the β-value of connectivity-based tau-PET rate of change) were associated with cognitive decline as measured by annual change on a cognitive composite score over time, designed to capture cognitive changes in the earliest AD stages (Fig. 4a–c). Given the observation that soluble p-tau increases early in AD and prior to the formation of neocortical insoluble tau aggregates, we then tested whether measures related to the accumulation rate of insoluble tau aggregates mediated the association between soluble p-tau concentrations and subsequent cognitive decline. First, the strength (β-value) of the association between accumulation rate of tau aggregates and functional connectivity to tau epicenters mediated 25% of the association between soluble p-tau and cognitive decline (Fig. 4f), suggesting a partial mediating effect of faster connectivity-based tau aggregates accumulation. Second, the accumulation rate of tau aggregates in the regions most connected to the subject-specific tau epicenter (Q1) mediated 60% of the association between soluble p-tau concentrations and the rate of cognitive decline (Fig. 4g). In sensitivity analyses, using the slopes of MMSE scores to measure cognitive decline, we found very similar mediating effects of tau aggregation (for mediating effects of the connectivity-based β-value: c’-c = −0.09 [95% CI −0.19, 0.00], p = 0.04; for mediating effect of the tau-PET rate of change in Q1: c’-c = −0.25 [−0.39, −0.13], p < 0.001). The same analyses were also repeated splitting the non-demented group into Aβ-positive CU and MCI separately. We observe a full mediation effect of tau-PET rate of change, either in Q1 or in the temporal meta-ROI between soluble p-tau and cognitive decline on the cognitive composite score in CU (c’-c = −0.21 [−0.40, −0.06], p = 0.002), and on decline on MMSE in MCI (c’-c = −0.24 [−0.46, −0.04], p = 0.01), see Supplementary Fig. 12 for detailed statistics. The only analysis that we could not replicate in individual groups was the mediating effect of the β-value of connectivity-based tau-PET change (Fig. 4f), which was also of smaller magnitude in the full group of non-demented participants. Taken together, the results indicate that higher concentrations of soluble p-tau are associated with cognitive decline in early AD, which is mediated by increased accumulation rates of insoluble tau aggregates.
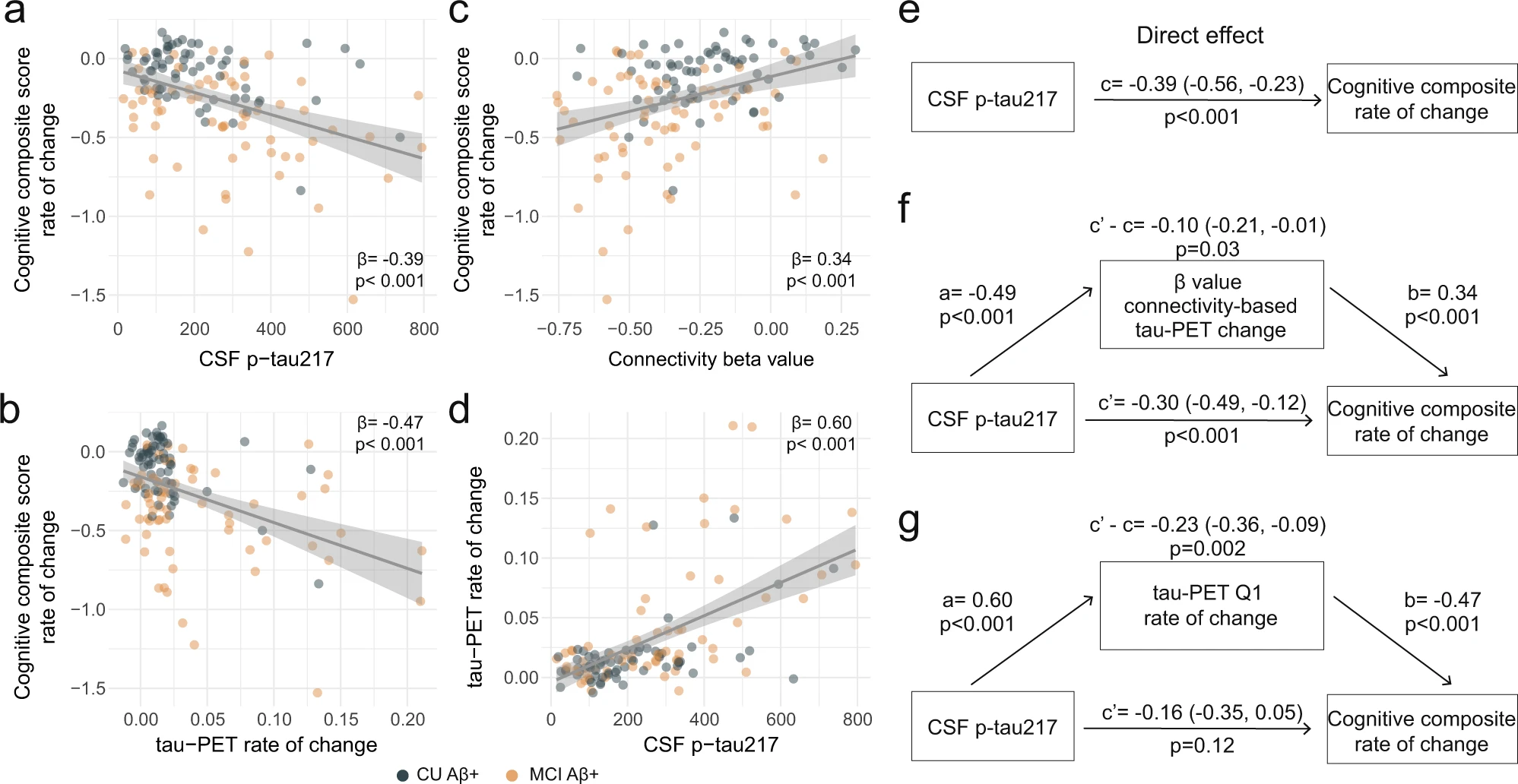
Tau aggregates accumulation mediates associations between CSF p-tau217 and cognitive decline in Aβ-positive non-demented participants.
a–d Scatter plots of associations relevant to subsequent mediation analyses, beta coefficients from linear regressions adjusting for age, sex and education are reported. a Association between CSF p-tau217 and rate of change on the cognitive composite score. b Association between tau-PET rate of change in Q1 and rate of change on the cognitive composite score. c Association between β-value from the correlation between tau-PET rate of change and connectivity-based distance to epicenters across all brain regions and rate of change on the cognitive composite score. d Association between CSF p-tau217 and tau-PET rate of change in Q1. e–g Mediation analysis of the relationship between CSF p-tau217, measures of tau-PET and cognitive decline measured as the rate of change on the cognitive composite score. The direct effect (c) of CSF p-tau217 on cognitive decline is shown in e. Analyses are shown with β-value based on connectivity and tau-PET change (f), and tau-PET rate of change in Q1 (g) as mediators. The mediated effect is designated c–c’. The remaining effect of CSF p-tau217 on cognitive decline after adjusting for the mediator is designated c′. 95% confidence intervals derived from 1000 simulations are reported in parentheses. The direct effect of CSF p-tau217 on the mediator is a, and the direct effect of the mediator on cognitive decline is b. The β-value based on connectivity and tau-PET change (f) and tau-PET rate of change (g) mediated the relationship between CSF p-tau217 and cognitive decline. To facilitate model comparisons, all models use continuous standardized (z-score) data for variables of interest. All linear regressions performed were two-sided, without adjustment for multiple comparisons and error bands correspond to the 95% confidence interval. Aβ beta-amyloid, CSF cerebrospinal fluid, PET positron emission tomography, p-tau phosphorylated tau, Q quartile.
Based on effect sizes determined in BioFINDER-2, sample sizes between 75 and 82 participants would have been needed to assess effects of soluble p-tau on cognitive decline in ADNI. Since only 28 participants had longitudinal cognitive assessments, this set of analyses was not conducted in ADNI.
Distinct associations between soluble p-tau and accumulation of tau aggregates in AD dementia
All previous analyses focused on non-demented participants, to study the effects of soluble p-tau and connectivity on tau-PET rate of change in early stages of AD. To investigate the full AD clinical continuum, we repeated the main analyses focusing on Aβ-positive individuals with AD dementia. Our motivation to focus on AD dementia patients separately was based on the plateau-phase reached by soluble p-tau species in more advanced disease stages (Fig. 5a with CSF Aβ42/40 to include the full sample since AD dementia patients do not undergo Aβ PET, and Supplementary Fig. 13 with Aβ PET in a restricted sample). In Aβ-positive non-demented participants, higher soluble p-tau concentrations were associated with faster tau aggregates accumulation, above levels of Aβ and baseline tau aggregates. In contrast, in AD dementia, soluble p-tau concentrations were not associated with the accumulation rates of tau aggregates in any region after adjusting for baseline levels of local tau aggregates (Fig. 5b). The expected negative association between greater regional accumulation rates of tau aggregates and connectivity to the tau epicenters across the brain was present at the dementia stage, but the strength of this association (β-value) was not related to soluble p-tau levels (Fig. 5c). Similarly, soluble p-tau concentrations did not have a direct effect on cognitive decline (Fig. 5d). Rather, the accumulation rate of tau aggregates over time was most associated with cognitive decline at this stage of the disease (Fig. 5e in Q1 and β = −0.36, p < 0.001 for association in the temporal meta-ROI). Further, soluble Aβ levels, measured as the CSF ratio of Aβ42/40, were not related to the accumulation of tau aggregates (β = 0.07, p = 0.55 for tau-PET rate of change in Q1) or cognitive decline (β = −0.21, p = 0.10 for MMSE slope). Overall, these results suggest that the accumulation rate of tau aggregates and cognitive decline seem independent of soluble p-tau concentrations in the dementia stage of the disease.
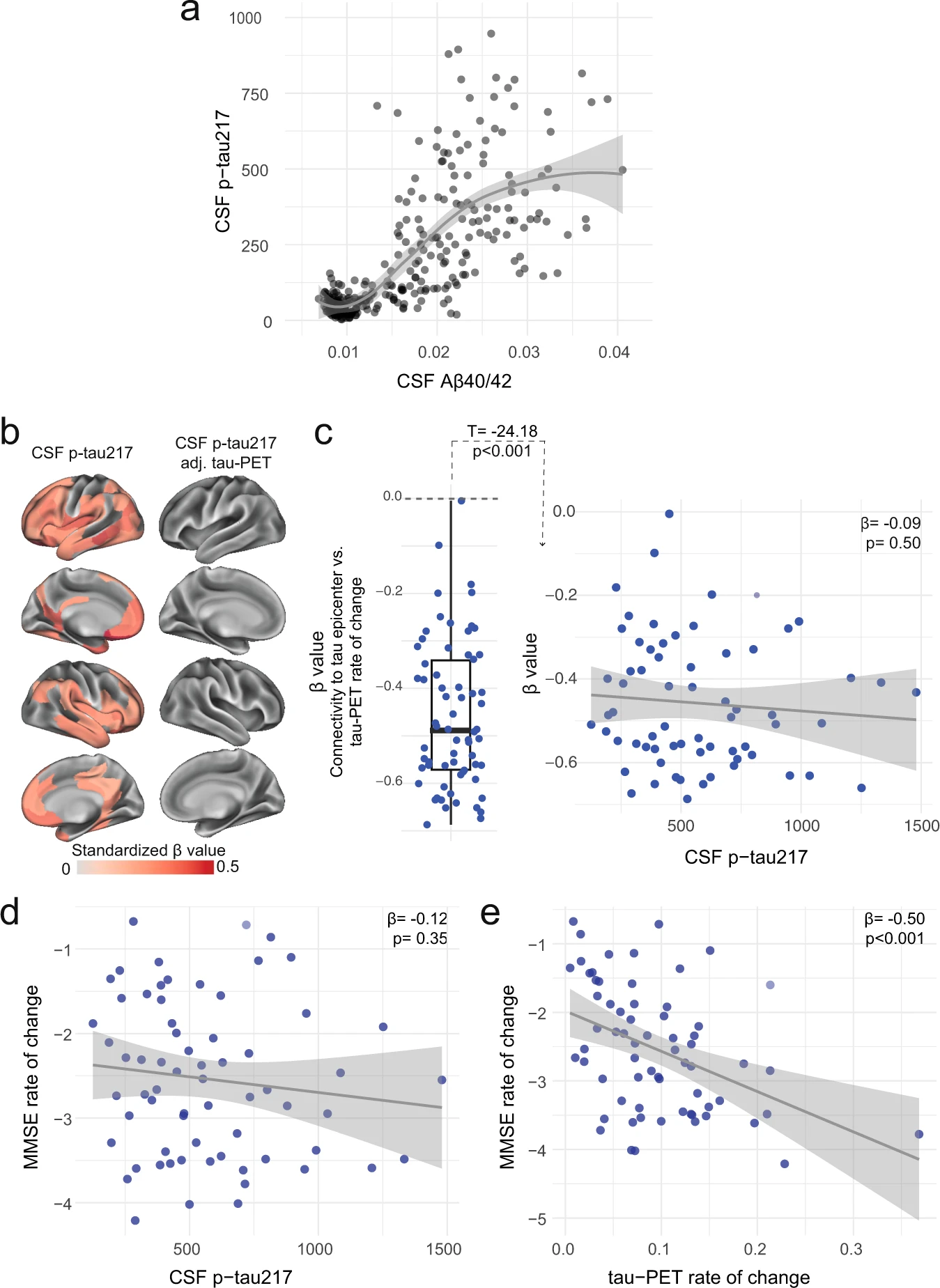
Distinct associations between CSF p-tau217 and tau-PET rate of change in the AD dementia stage.
a Associations between the ratio of CSF Aβ40/42 from Elecsys and CSF p-tau217 across the AD continuum. CSF Aβ40/42 is shown here instead of PET to include the entire BioFINDER-2 cohort, since AD dementia patients do not undergo Aβ-PET. A similar association with Aβ-PET SUVR is shown in a restricted sample without AD dementia patients in Supplementary Fig. 13. The association is nonlinear and flatten at high Aβ load. b CSF p-tau217 alone was mildly associated with tau-PET rate of change (left column), which was not the case when additionally adjusting for regional baseline tau-PET SUVR (right column). c Box plot showing the expected negative β-value from the correlation between tau-PET rate of change and connectivity-based distance to epicenters across all AD patients, but this β-value was not related to CSF p-tau217 levels. Scatter plots of associations between CSF p-tau217 (d) and tau-PET rate of change in Q1 (e) and cognitive decline, as measured by MMSE rate of change. Beta coefficients from linear regressions adjusting for age, sex and education are reported. The rate of tau aggregates accumulation was related to cognitive decline, which was not the case for CSF p-tau217. All linear regressions performed were two-sided, without adjustment for multiple comparisons and error bands correspond to the 95% confidence interval. Aβ beta-amyloid, AD Alzheimer’s disease, CSF cerebrospinal fluid, MMSE Mini-mental state examination, PET positron emission tomography, p-tau phosphorylated tau.
Discussion
The major aim of the present longitudinal biomarker study was to assess the relations between soluble p-tau concentrations and Aβ-related accumulation of insoluble tau aggregates over time in AD. In a large sample of Aβ-positive non-demented individuals of the BioFINDER-2 cohort, we found first, that soluble CSF p-tau fully mediated the effects of regional Aβ on the subsequent accumulation rate of tau aggregates measured with longitudinal tau-PET, and second, that elevated soluble p-tau was associated with faster accumulation of tau aggregates across functionally connected brain regions. Importantly, these findings were replicated in the ADNI cohort. Third, we found that elevated soluble p-tau concentrations were associated with faster cognitive decline in early stages of AD, which was mediated by faster accumulation rates of tau aggregates. All findings in non-demented group also even held when analyzing CU and MCI groups separately (Supplementary Figs. 6, 8 and 12). However, soluble p-tau concentrations, that plateau at the late-stage of Aβ accumulation, were no longer associated with local accumulation of tau aggregates or cognitive decline in patients with AD dementia. At this advanced stage, baseline levels of tau aggregates predicted subsequent accumulation of tau aggregates in the same brain regions37, and we further showed that tau aggregation rates related to greater cognitive decline. These results suggest a close link between the accumulation of tau aggregates and clinical deterioration once Aβ and p-tau levels have plateaued in late-stage AD. Taken together, these findings are congruent with the hypothesis that Aβ-induced soluble p-tau concentrations might play a key role in initiating the aggregation and connectivity-mediated accumulation of tau pathology in early-stage AD and that local tau seeding and self-replication predominate once soluble p-tau concentrations have plateaued in AD dementia. This might have implications for clinical trials, since drugs reducing soluble p-tau concentrations (like anti-Aβ therapies or genetic anti-tau treatments) may be promising therapeutic strategies to prevent further accumulation and spread of tau aggregates and cognitive decline in early stages of AD. On the other hand, during the dementia stage of AD, directly targeting the local tau aggregates might prove to be more adequate than targeting soluble p-tau species or Aβ.
To gain a better understanding of AD pathogenesis, it is important to elucidate how key biomarkers relate to longitudinal changes along the disease continuum. Here we found that soluble p-tau was a key biomarker in the Aβ-related accumulation of tau aggregates and of cognitive decline over time in early AD. Importantly, the effect of CSF p-tau on increases in tau-aggregates survived adjustment for baseline levels of Aβ and tau aggregates in the same brain region, suggesting a unique contribution of soluble p-tau in subsequent tau aggregation, and not only on baseline levels of tau aggregates38,39. Moreover, soluble p-tau mediated the association between Aβ and accumulation rate of subsequent tau aggregates, expanding on previous cross-sectional results7,10. As such, Aβ-related increases in soluble p-tau may be a key initial step in the Aβ cascade that determines the accumulation of aggregated tau pathology, and thereby leading to faster cognitive decline in early AD, and this cascade of events was even found in the preclinical stage of the disease. This finding expands on previous studies linking longitudinal tau-PET and cognitive decline40,41, by highlighting the importance of soluble p-tau in early stages of the disease.
From a pathophysiological point of view, Aβ has been shown to trigger increased synthesis, hyperphosphorylation and secretion of tau proteins from neurons, which leads to elevated interstitial p-tau concentrations, that pass into CSF and blood plasma where it can be detected in vivo using biomarkers1,9. Preclinical studies provide strong support for neuronal activity as a putative link between Aβ and tau secretion and accumulation. Aβ has consistently been shown to induce a hyper-excitatory shift in neuronal activity21,42,43, and this neuronal hyperexcitability may then induce increased p-tau secretion and subsequent spreading, with neuronal tau secretion being drastically enhanced by elevated neuronal activity20,23,24. This view is supported by our result showing that elevated soluble p-tau was associated with faster accumulation of tau aggregates across functionally connected brain regions. While accumulation of tau aggregates in a given brain region was related to its functional connectivity to tau epicenters27,36,44, the accumulation of tau aggregates from local epicenters to connected regions was enhanced in the face of abnormal soluble p-tau. Overall, both connectivity and soluble p-tau were important factors of the rate of tau aggregates accumulation. These findings suggest that Aβ-related soluble p-tau increases could be key prerequisite for the expansion of tau aggregates across functionally connected brain regions in early AD.
Importantly, the results described above only apply to early pre-dementia AD stages and not to the dementia stage of AD. We found that the key role of soluble p-tau in the accumulation process of tau aggregates was no longer observed in patients with AD dementia. This can likely be explained by a plateau stage of Aβ aggregates and soluble p-tau levels occurring in more advanced disease stages10,45. In our data, soluble p-tau levels had a minor effect on further accumulation of tau aggregates and connectivity-based tau spreading in AD dementia, and only the tau aggregatse accumulation rate was associated with cognitive decline (and not p-tau levels).
The main strength of this study is the integration of cross-sectional and longitudinal fluid and neuroimaging biomarkers of AD pathology and longitudinal cognitive measures across the clinical spectrum of AD. There are also several limitations. First, although we are expanding on previous studies that were limited by shorter follow-up or smaller sample sizes46,47,48, a longer follow-up time of tau-PET would increase the rate of tau aggregates accumulation in earlier stages of AD, which span many years. The correlational study design precludes us from making conclusive statements on soluble p-tau as the main driver of subsequent tau aggregation, but investigating factors related to tau-PET rate of change brings novel insight into AD pathophysiology in vivo. Second, there were some differences between the biomarkers and cognitive data between BioFINDER-2 and ADNI. In BioFINDER-2 the focus was on CSF p-tau217, having shown greatest association with Aβ12,49, but the main results could still be validated in ADNI where only CSF p-tau181 was available. Unfortunately, cognitive decline could not be investigated in ADNI due to the small number of participants with longitudinal cognitive data, but we showed the robustness of these results in BioFINDER-2 by using two cognitive scores and different regions of interest of tau aggregates accumulation. Lastly, we acknowledge that functional connectivity is an indirect measure of brain activity, but still has been shown to be related to mono- and polysynaptic pathways50.
In conclusion, reconciling both early and later stages of the disease, we propose an integrative model of how Aβ fibrils, soluble p-tau concentrations, functional connectivity, accumulation rates of insoluble tau aggregates and cognitive decline are interrelated across the entire clinical continuum of AD (Fig. 6). In this model, initially the Aβ-related increase of p-tau seeds are taken up by neurons to initiate the misfolding and aggregation of tau in a given brain region, with more substrate (i.e. soluble p-tau) for aggregation leading to more rapid local tau aggregation. Later on, local self-replication and accumulation of misfolded tau aggregates may take over once a critical threshold of tau aggregates is reached. This distinct-stage process also aligns with recent evidence of trans-neuronal tau spreading being critical for the initial expansion of tau pathology, but local replication of tau pathology driving accumulation of tau aggregates in later stages51. Stemming from this model, our work has potential implications for therapeutic approaches. We propose that targeting Aβ fibrils and soluble p-tau in early AD may be a promising strategy to slow the formation of tau aggregates, and thereby to preserve cognitive abilities for a longer period. Preliminary evidence from recent phase 2 and 3 clinical trials suggest that anti-Aβ therapies promptly reduce soluble p-tau concentrations in both CSF and plasma, reenforcing the close relationship between Aβ pathology and increases in extracellular p-tau levels52,53,54. Applying the same principles to anti-tau therapies (see ref. 55 for a review), we speculate that approaches reducing tau production and phosphorylation (e.g. antisense oligonucleotides, post-translational modification modulators, passive immunotherapies) would be most effective early on. On the other hand, in late stages of the disease, when local tau aggregation is the predominant pathway leading to tau accumulation, targeting tau aggregates may exert stronger clinical benefits. As such, it is possible that therapeutics acting on tau aggregation inhibition and active clearance of aggregates might be more beneficial in the dementia stage. Our findings may be a starting point for future precision-medicine interventions that target the dominant pathway that determines tau aggregation, which are Aβ and soluble p-tau increases in early AD and tau aggregates in late AD.
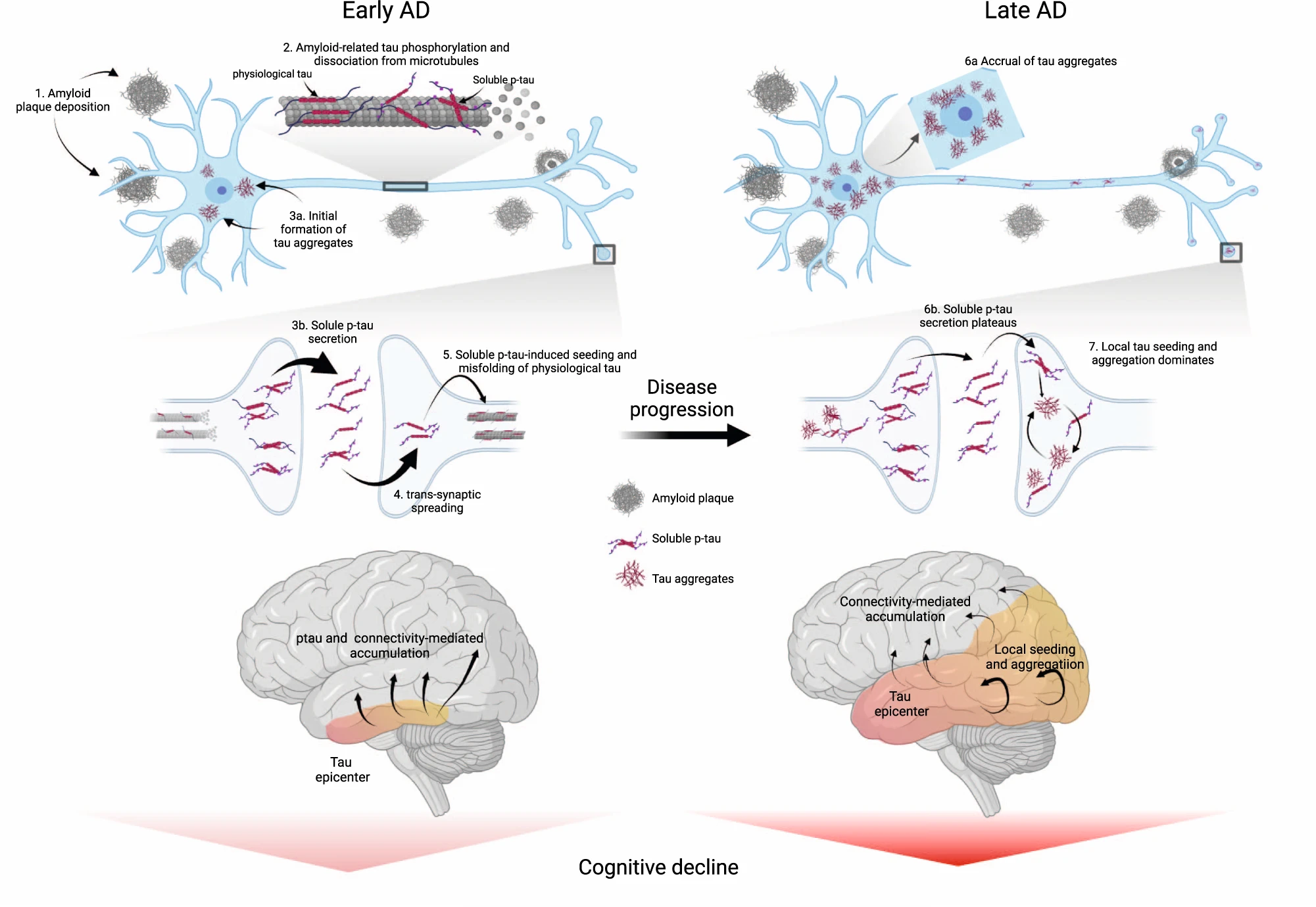
Proposed model of tau pathology accumulation in Alzheimer’s disease.
In early AD, soluble or insoluble Aβ triggers increased concentrations and secretion of soluble p-tau, followed by post-synaptic uptake of p-tau seeds that lead to tau misfolding and aggregation. In late stages of the disease, when soluble p-tau concentrations have reached a plateau, local tau aggregates rather dominate in driving further local tau aggregation. Figure created with BioRender.com. Aβ beta-amyloid, AD Alzheimer’s disease, p-tau phosphorylated tau.