
Adenosine A2A receptors control synaptic remodeling in the adult brain

By Xinli Xu, Rui O. Beleza, Francisco Q. Gonçalves, Sergio Valbuena, Sofia Alçada-Morais, Nélio Gonçalves, Joana Magalhães, João M. M. Rocha, Sofia Ferreira, Ana S. G. Figueira, Juan Lerma, Rodrigo A. Cunha, Ricardo J. Rodrigues, and Joana M. Marques
Excerpt from the article published in Scientific Reports 12, 14690, 29 August 2022, DOI: https://doi.org/10.1038/s41598-022-18884-4
Editor’s Highlights
- The current knowledge on the mechanisms driving circuit re-wiring in the mature central nervous system is limited.
- An example of such re-wiring processes in the adult brain is the hippocampal mossy fiber (MF) sprouting found in patients and in animal models of temporal lobe epilepsy (TLE).
- Other disorders displaying also circuit alterations are neuropsychiatric disorders affecting adults and prevalent on ageing.
- Adenosine A2A receptors (A2AR) control axon formation/outgrowth and synapse stabilization during development.
- The overactivation of A2ARs is a molecular mechanism underlying synaptic remodeling in the injured brain, namely SE-induced hippocampal MF sprouting.
- The A2AR-mediated mechanism seems intrinsic to neurons rather than involving astrogliosis and inflammation, which are also characteristic features of epilepsy that are associated with the development of seizures and with the ability to contribute to synaptic remodeling.
- A2ARs contribute to epilepsy-related circuit remodeling, which may involve the reactivation of the ability of A2ARs to control axon formation and outgrowth during development.
Abstract
The molecular mechanisms underlying circuit re-wiring in the mature brain remains ill-defined. An eloquent example of adult circuit remodelling is the hippocampal mossy fiber (MF) sprouting found in diseases such as temporal lobe epilepsy. The molecular determinants underlying this retrograde re-wiring remain unclear. This may involve signaling system(s) controlling axon specification/growth during neurodevelopment reactivated during epileptogenesis. Since adenosine A2A receptors (A2AR) control axon formation/outgrowth and synapse stabilization during development, we now examined the contribution of A2AR to MF sprouting. A2AR blockade significantly attenuated status epilepticus(SE)-induced MF sprouting in a rat pilocarpine model. This involves A2AR located in dentate granule cells since their knockdown selectively in dentate granule cells reduced MF sprouting, most likely through the ability of A2AR to induce the formation/outgrowth of abnormal secondary axons found in rat hippocampal neurons. These A2AR should be activated by extracellular ATP-derived adenosine since a similar prevention/attenuation of SE-induced hippocampal MF sprouting was observed in CD73 knockout mice. These findings demonstrate that A2AR contribute to epilepsy-related MF sprouting, most likely through the reactivation of the ability of A2AR to control axon formation/outgrowth observed during neurodevelopment. These results frame the CD73-A2AR axis as a regulator of circuit remodeling in the mature brain.
Introduction
The current knowledge on the mechanisms driving circuit re-wiring in the mature central nervous system is limited. Yet, this knowledge is fundamental to understand and correctly promote nerve regeneration, control stem cell grafting or adult-born neurons integration or prevent pathological circuit remodeling. A very well-established example of such re-wiring processes in the adult brain is the hippocampal mossy fiber (MF) sprouting found in patients and in animal models of temporal lobe epilepsy (TLE)1,2.
The retrodirective growth of MF into the inner molecular layer of the dentate gyrus (DG) 3,4seems to be due to a dysregulation of guidance cues involved in the establishment of normal MF connectivity during development5,6,7,8 but the underlying molecular determinants are still unclear. BDNF has been associated to this initial axonal branching9,10,11 but other studies observed MF sprouting in the absence of BDNF12 or showed that BDNF is not sufficient to induce MF sprouting13,14. which critically requires abnormal circuit activity15.
One unexplored set of candidates to regulate mature circuit remodeling, in particular hippocampal MF sprouting, are purines; purines are released in an activity-dependent manner16 and can control neuronal migration and wiring during development, particularly through adenosine A2A receptors (A2AR). ATP is preferentially released from MF terminals at high-frequency stimulations17,18 and we recently reported that A2ARs control axon formation and outgrowth of migrating cortical projection neurons during embryogenesis19. being also involved in synaptic stabilization during development20. Besides, A2ARs are located in hippocampal MF terminals18. contribute to synaptic failure and transient decrease in cell proliferation in the DG upon oxygen–glucose deprivation21. are active during epileptogenesis18,22,23,24 and their pharmacological blockade or genetic deletion prevent seizure-induced neurodegeneration23 and attenuate the progressive seizure severity in different models of kindling23,25.
In this study, we provide evidence that A2ARs, in particular those located in dentate granule cells are activated by ATP-derived adenosine and contribute to status epilepticus (SE)-induced hippocampal MF sprouting, most likely by inducing axon formation and outgrowth. This demonstrates a reactivation of adenosine-induced A2AR-mediated control of brain wiring in the adult brain, impacting on circuit remodeling after SE.
Results
Pharmacological activation of A2ARs induces the formation of secondary aberrant axons in cultured hippocampal neurons
To start addressing if A2ARs control axon formation and/or outgrowth of hippocampal neurons, as previously observed in cortical neurons19 we first evaluated the impact of the pharmacological manipulation of A2ARs in axon formation/outgrowth of cultured E18-rat derived hippocampal neurons. We observed that the pharmacological activation of A2ARs with the selective agonist CGS21680 (30 nM) from DIV0 caused an increase in the number of axons per neuron, reflecting an increase in the percentage of neurons with more than one axon, analyzed at DIV3 (Fig. 1a,b). CGS21680 also increased axonal length without affecting axonal branching (Fig. 1c). The selective antagonist of A2AR, SCH58261 (50 nM), did not significantly modify neither axon formation nor axonal elongation (Fig. 1b,c), showing that the tonic activation of A2ARs by endogenous adenosine is absent or does not affect axon formation/outgrowth. Accordingly, the knockdown of A2ARs, using a validated short-hairpin RNA expressing a construct against A2AR (shRNA-A2AR), neither significantly modified the number of axons per neuron nor axon elongation, analyzed at DIV3 (Fig. 1d and Supplementary Figs. 1, 2). The increase in the number of axons per neuron and axon length caused by CGS21680 are mediated by the activation of A2ARs since it was not observed in the presence of SCH58261 (Fig. 1b,c) and was observed in cells electroporated with a short-hairpin RNA expressing a construct against a non-targeting control (shRNA-Control) but not in cells expressing shRNA-A2AR (Fig. 1d).
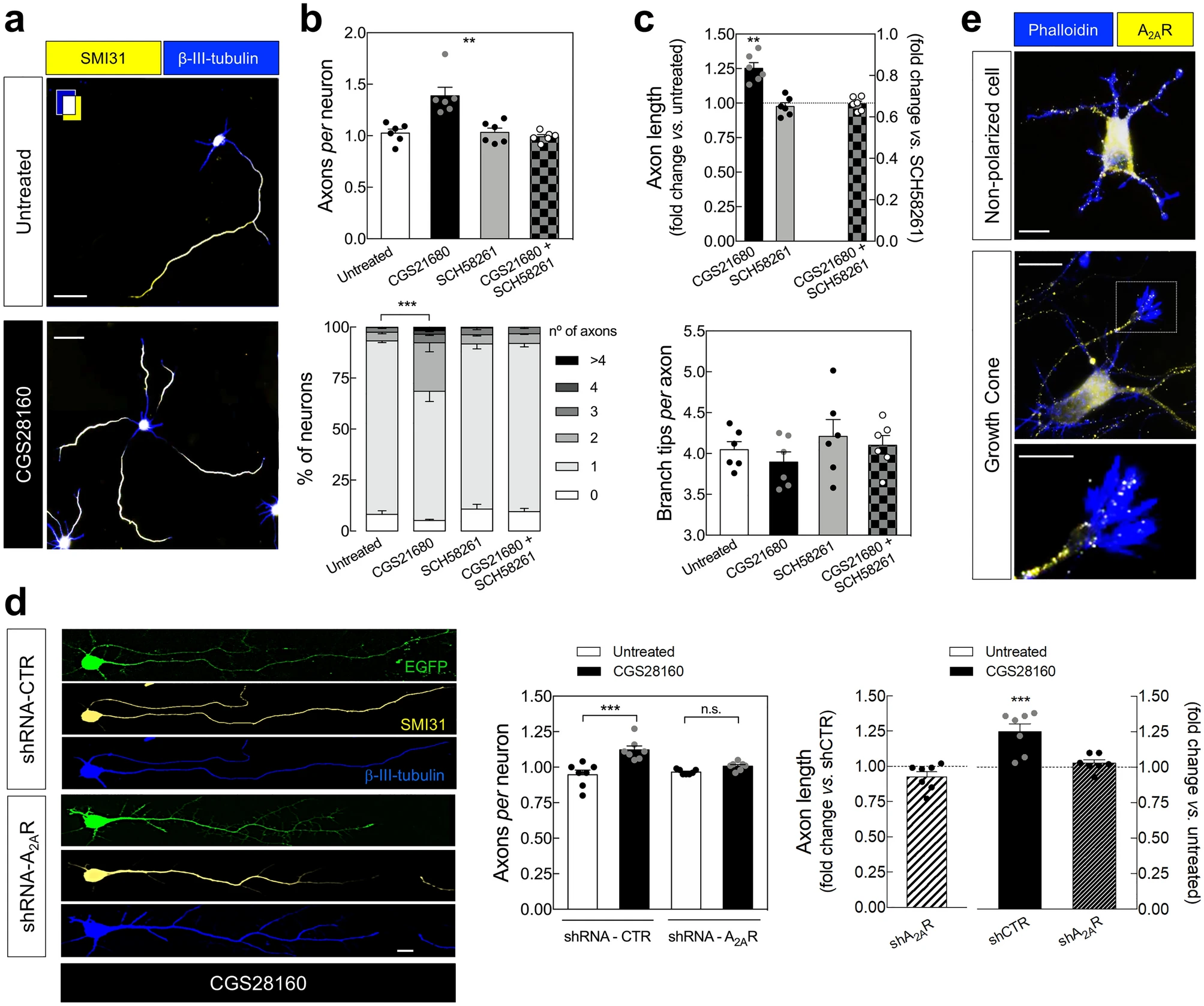
Pharmacological activation of A2ARs induces the formation of abnormal secondary axons in cultured hippocampal neurons. (a) Representative images of E18 rat-derived hippocampal neurons cultured in the absence and in the presence of the selective agonist of A2ARs, CGS21680 (30 nM), from DIV0, double immunolabeled with SMI31 (axonal marker; yellow) and βIII-tubulin (neuronal marker; blue) antibodies at DIV 3 (scale bar, 30 µm), showing that (b) the exposure to CGS21680 increased the number of axons per neuron (upper graph), reflecting an increased percentage of neurons with multiple axons (lower graph). The selective antagonist of A2ARs, SCH58261 (50 nM), did not modify the average number of axons per neuron, but prevented the ability of CGS21680 to modify the number of axons per neuron. (c) CGS21680 increased axonal length (in those cells displaying more than one axon, it was counted the longest axon), whereas SCH58261 was devoid of effects. In the presence of SCH58261, CGS21680 did not modify axonal length. Neither CGS21680 nor SCH58261 significantly modified axonal branching. Data are mean ± SEM quantified from 6 independent cultures, analyzing a minimum of 100 cells perculture and condition. (d) In cells electroporated at DIV0 with either shRNA-Control (shCTR) or shRNA-A2AR (shA2AR) (EGFP+), CGS21680 increased the number of axons per neuron and axonal length in cells transfected with shCTR but not in cells transfected with shA2AR. Scale bar, 10 μm. Data are mean ± SEM quantified from 7 independent cultures, analyzing a minimum of 25 transfected cells per culture and condition. **P < 0.01 and ***P < 0.001. (b upper; clower) one-way ANOVA with Dunnet’s post-hoc test; (b lower; d left) two-way ANOVA with Sidak’s post-hoc test; (c upper; d right) one-sample t-test vs.hypothetical value of 1. (e) Immunocytochemical analysis of A2AR immunoreactivity (yellow) in a non-polarized neuron and in an axonal growth cone labelled with phalloidin (blue). Scale bar for upper and middle image, 10 μm; scale bar for the lower image, 5 μm.
These results show that the pharmacological activation of A2ARs promotes axonal elongation and induces the formation of secondary abnormal axons in developing rat hippocampal neurons. Accordingly, we observed an immunoreactivity for A2AR in non-polarized cells as well as in axonal growth cones (Fig. 1e). These findings in hippocampal neurons converge with a previous report in cortical neurons19, suggesting that A2ARs control axogenesis in different neuronal populations.
A2ARs contribute to status epilepticus-induced hippocampal mossy fiber sprouting
Hippocampal organotypic slices display spontaneous MF sprouting in the molecular layer of the DG26,27,28 supporting an enhanced excitation of dentate granule cells29. In hippocampal organotypic slices prepared from P5-7 mice, stimulation of MF at DIV7 elicited synaptic currents in dentate granule cells (Fig. 2). In slices cultured in the presence of the A2AR antagonist SCH58261 (50 nM), the current density was not significantly modified (Fig. 2), indicating that the formation of these recurrent excitatory inputs at the dentate granule cells in hippocampal organotypic slices is unrelated to the activation of A2ARs by endogenous adenosine.
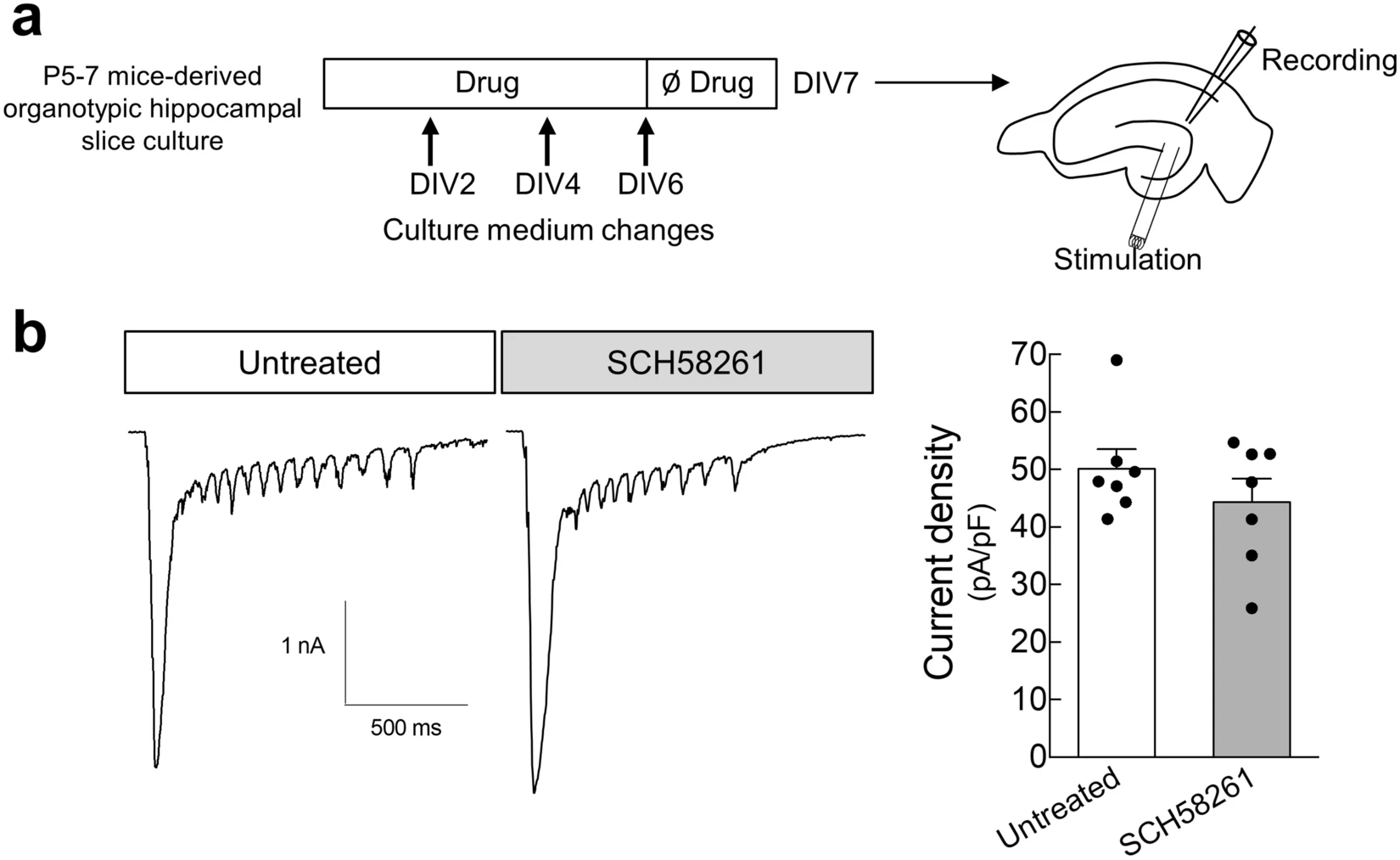
Pharmacological blockade of A2ARs does not modify the spontaneously-formed mossy fiber recurrent pathways in hippocampal organotypic slices. (a) P5-7 mice-derived hippocampal organotypic slices were cultured either in the absence or in the presence of SCH58261 (50 nM) until DIV 6, and at DIV 7 synaptic currents at dentate granule cells elicited by stimulation of mossy fibers at the hilus were recorded by whole-cell patch-clamp. (b) SCH58261 did not significantly modify the density of synaptic current in dentate granule cells upon stimulation at the hilus, as depicted in the representative traces and summarized in the histogram. Data are mean ± SEM quantified from 7 independent cultures, recording 6–13 cells per culture per condition. P > 0.05 unpaired t-test.
Next, we evaluated the contribution of A2ARs to hippocampal MF sprouting induced by SE. We resorted to the pilocarpine-induced model of SE in rats, which triggers a robust hippocampal MF sprouting within weeks to months after SE30. Forty-two days after SE induction, a positive Timm staining demonstrated MF sprouting in the granule cell and molecular layers of the DG (Fig. 3a,b). Only pilocarpine-treated animals reaching at least stage 4 seizures were considered in this study. To appraise the contribution of A2ARs, rats were injected daily with SCH58261 (0.1 mg/kg, i.p.) or vehicle, beginning 10 h after the onset of SE (Fig. 3a). The extent of MF sprouting was assessed by estimating the percentage of the granule cell layer plus molecular layer that presented Timm staining (Fig. 3b). In control rats that had not experienced SE, non-significant levels of Timm staining were observed in the granule cell layer and inner molecular layer either in vehicle or SCH58261-treated rats (Fig. 3c,d). In contrast, animals that had developed SE and had been administered with vehicle for 42 days, displayed a significantly denser Timm staining in the entire granule cell layer and inner molecular layer of the DG (P < 0.0001). In rats administered with SCH58261, SE still induced a significantly higher Timm staining than in control (P = 0.013 SE-SCH58261 vs.Control-SCH58261), but considerably lower than the observed in vehicle-injected rats subjected to SE (P = 0.039 for SE vs. SCH58261 interaction; P = 0.004 for SE-vehicle vs. SE-SCH58261) (Fig. 3c,d). SE-induced MF sprouting was observed along the entire septotemporal axis (Fig. 3e) both in control and, albeit with significantly lower levels, in SCH58261-treated animals (Fig. 3e).
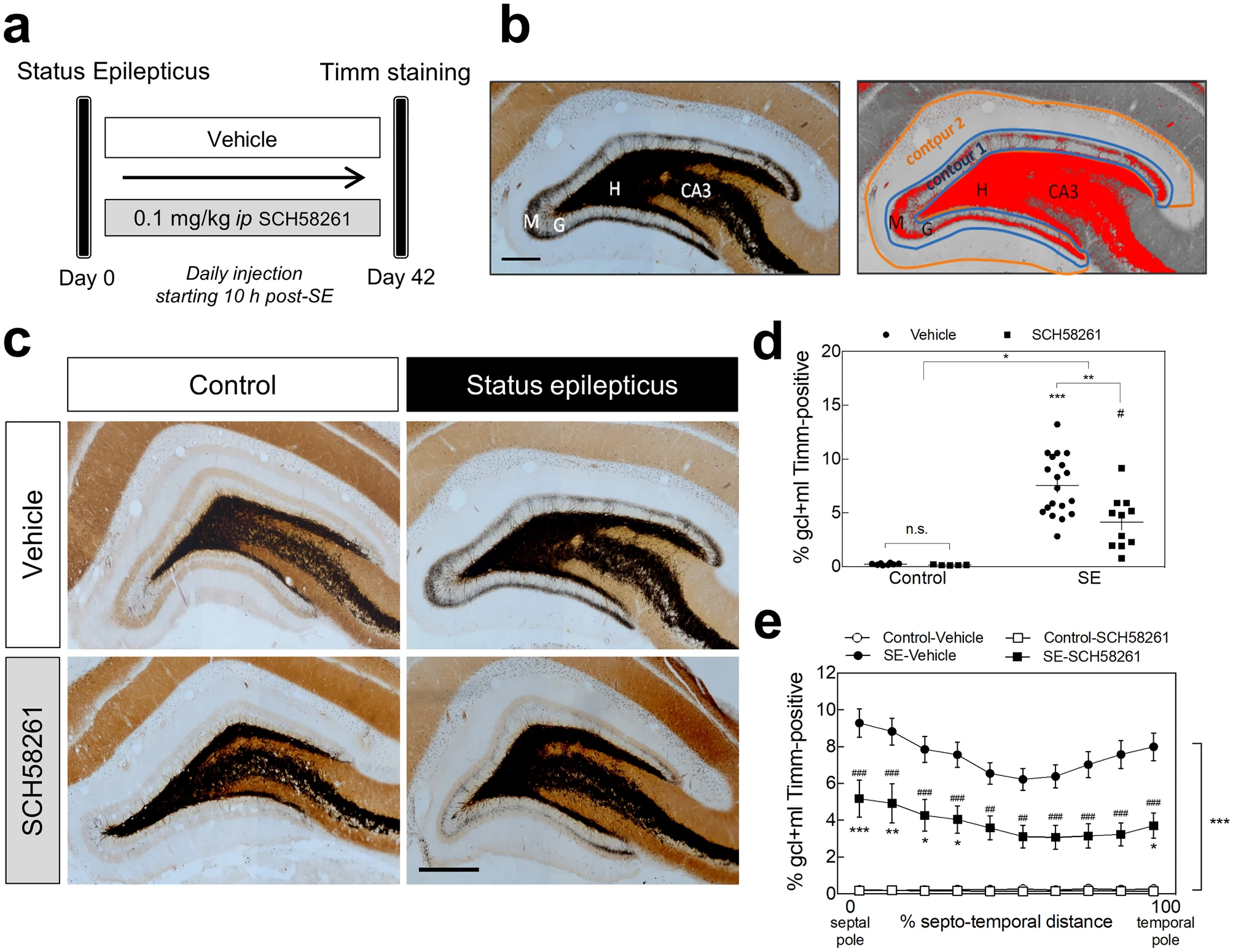
Pharmacological blockade of A2ARs reduces status epilepticus-induced mossy fiber sprouting in the rat hippocampus. (a) Six/seven weeks old rats were either injected with saline (Control group) or with pilocarpine (Status epilepticus group—SE), 30 min after administration of scopolamine methylbromide (2 mg/kg, i.p.). Only animals reaching at least stage 4 seizures were considered in this study. Convulsions were suppressed with diazepam (10 mg/kg, i.p.) 2 h after the occurrence of the first stage 4 seizure. The rats from each group were assigned into two subgroups: one subgroup was daily injected with the selective antagonist of A2ARs, SCH58261 (0.1 mg/kg i.p.), and the other with vehicle (0.2% Tween-20 in saline), starting 10 h after the onset of status epilepticus (SE). (b) Forty-two days later, hippocampal mossy fiber (MF) sprouting was analyzed by Timm staining as previously described102,103. The extent of MF sprouting was assessed by estimating the Timm-positive area (adjusting a darkness threshold setting until the selected area in red matched the black Timm-positive area of the same section)/volume measured in granule cell layer (GCL) + inner molecular layer (IML) (Contour 1) over the total volume/area of the GCL + molecular layer (ML) (Contour 2). H-Hilus; G-granule cell layer; M-molecular layer. (c) Rats that had developed SE displayed a dense Timm staining in the IML of the DG in comparison with Control animals. This staining was significantly lower in SE rats daily injected with SCH58261 as depicted in the representative images (scale bar, 300 μm) and (d) quantitatively summarized in the histogram. (e) The reduction in MF sprouting in SCH58261-treated SE rats was observed along the entire septotemporal axis of the DG. Control-Vehicle, n = 8; Control-SCH58261, n = 5; SE-Vehicle, n = 20; SE-SCH58261, n = 11. The data are mean ± SEM of the percentage of the GCL + ML displaying Timm staining. In (d), two-way ANOVA with Sidak’s post-hoctest; *P < 0.05, **P < 0.01, ***P < 0.001 and #P < 0.05 for SE-SCH58261 vs. Control-SCH58261. In (e), two-way ANOVA with Sidak’s post-hoc test; *P < 0.05, **P < 0.01 and ***P < 0.0001 for SE vs. Control either with vehicle or SCH58261; ##P < 0.01 and ###P < 0.001 for SCH58261 vs. vehicle with or without SE.
These results indicate that A2ARs contribute to SE-induced hippocampal MF sprouting.
A2ARs expressed by dentate granule cell neurons contribute to mossy fiber sprouting
If the contribution of A2ARs to SE-induced hippocampal MF sprouting is due to a direct control of axon formation/outgrowth, as observed in cultured hippocampal neurons, A2ARs expressed in dentate granule cells should mediate this effect. Thus, we evaluated the impact of knocking down A2ARs in dentate granule cells using lentivectors encoding the reporter gene EGFP and either shRNA-A2AR or shRNA-Control (Refs 31, 32 and Supplementary Figs. 1, 2).
Each rat was stereotaxically injected into the left or right DG with either shRNA-Control or shRNA-A2AR and was allowed to recover for 10 days before SE induction (Fig. 4a). Only animals reaching at least stage 4 seizures were considered in this study, and they were sacrificed 56 days after SE for the evaluation of MF sprouting in the inner molecular layer. This was performed by immunohistochemical identification of sprouted MF puncta by immunolabelling synaptoporin (Fig. 4b), a synaptic vesicle membrane protein highly enriched in MF terminals33,34,35. The lentivectors used have been previously shown to have a limited spread within the brain parenchyma36 infecting only neurons32. Accordingly, EGFP+-cells were all endowed with NeuN+-immunolabelling (Fig. 4c).
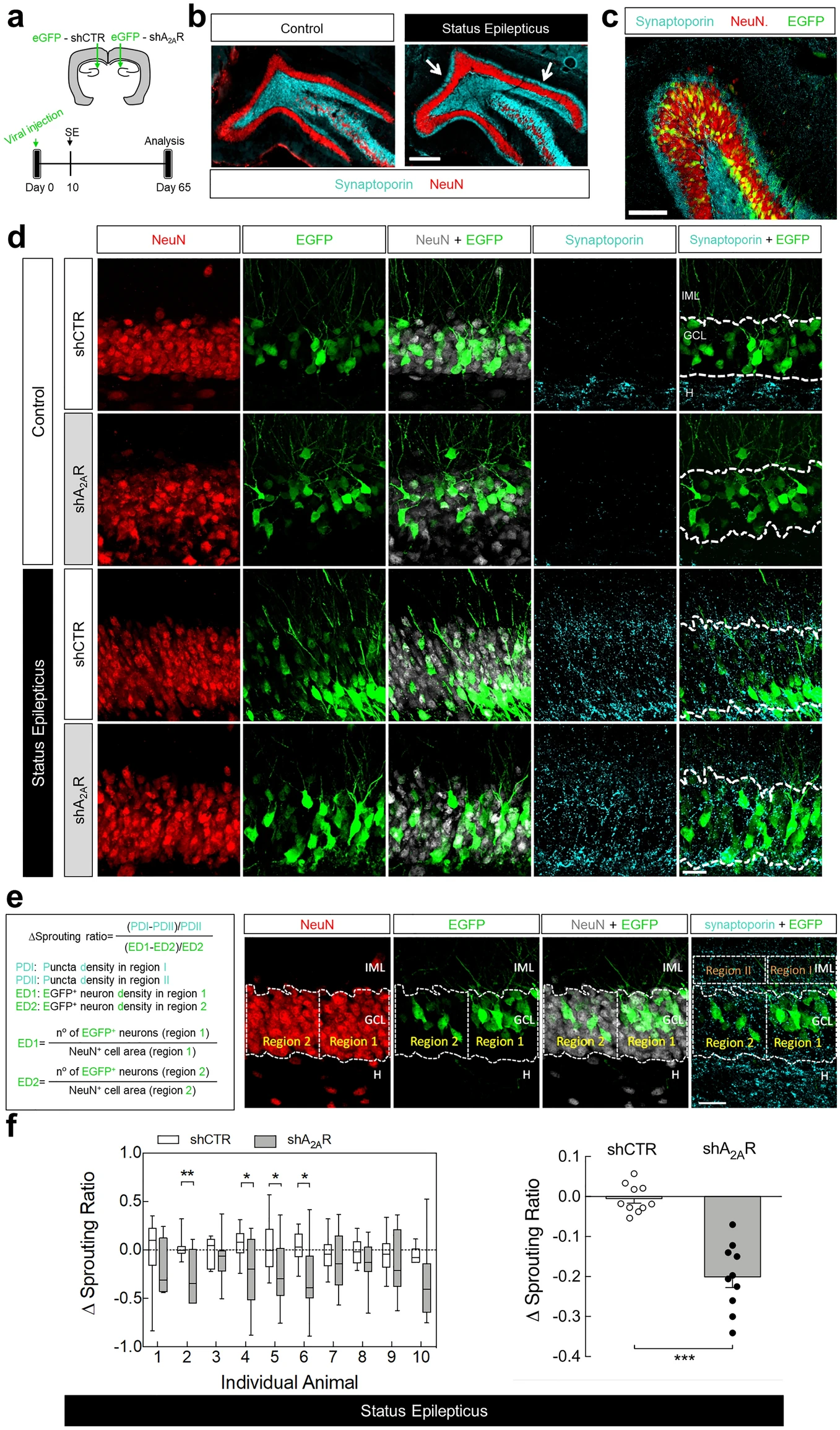
Knockdown of A2ARs in dentate granule cells reduces status epilepticus-induced hippocampal mossy fiber sprouting. (a) Lentivirus encoding EGFP and shRNA-Control (shCTR) or shRNA-A2AR (shA2AR) were injected into the hippocampal dentate gyrus (DG) in the left or in the right hemisphere. After 10 days, rats were injected with saline (Control group) or pilocarpine (Status epilepticus group). All rats were administered with scopolamine methylbromide (2 mg/kg, i.p.) 30 min before. Only animals reaching at least stage 4 seizures were considered. (b) After 55 days, mossy fiber (MF) sprouting was assessed by immunohistochemical analysis of synaptoporin (MF terminals marker). Representative images of the immunolabelling of synaptoporin and neuronal nuclei (NeuN) showing that in rats that experienced status epilepticus (SE) it was observed synaptoporin immunoreactivity in the inner molecular layer (IML) of the DG (white arrows), which was not observed in control rats. Scale bar, 300 μm. (c) Representative image showing a DG segment infected with lentivirus encoding shCTR from a rat that experienced SE, immunolabelled with synaptoporin and NeuN and displaying EGFP+-dentate granule cells. Scale bar, 100 µm. (d) Schematic illustration of the quantitative method used to analyse the extent of MF sprouting. A Δsprouting ratio for each acquired image with obviously different EGFP+-cell density in adjacent areas was calculated by measuring the incremental proportion of the synaptoporin (cyan) puncta density (PD2 to PD1) in the IML vs. the incremental proportion of EGFP+-neuron density (ED2 to ED1) in the granule cell layer (GCL), defined by NeuN immunoreactivity. (e) The brain sections from shCTR- or shRNA-A2AR-injected DG from control animals displayed no synaptoporin staining (cyan) in the IML. In SE group, shA2AR-injected DG sections displayed lower synaptoporin puncta densities in the IML region corresponding to the higher EGFP+-cell density region in GCL which was not observed in shCTR-injected DG. Scale bar, 25 μm. (f) Δsprouting ratio in shA2AR-injected DG and shCTR-injected DG in each individual animal that had experienced SE (left), supporting a significant lower Δsprouting in shA2AR-injected DG vs. shCTR-injected DG (right). The data are median and interquartile range (left graph) or mean ± SEM (right graph). *P < 0.05 and ***P < 0.001, paired t-test. H-Hilus.
Control animals showed no MF sprouting, evidenced by the lack of synaptoporin staining in the inner molecular layer, either in the DG injected with shRNA-Control or shRNA-A2AR (Fig. 4d). The rats that experienced SE displayed synaptoporin labelling in the granule cell and inner molecular layers (Fig. 4d). To estimate the impact of the knockdown of A2ARs in dentate granule cells in MF sprouting, we determined a Δsprouting ratio to evaluate the correlation between the density of infected granule cells (EGFP+) and sprouted MF puncta density in the adjacent inner molecular layer for each image acquired from animals that had experienced SE (Fig. 4e). We found a negative correlation between the density of synaptoporin puncta in the inner molecular layer and granule cell layer and EGFP+-cell density in the adjacent granule cell layer region in shRNA-A2AR-injected DG, but not in shRNA-Control-injected DG (Fig. 4e,f). In the analysis of individual animals, the Δsprouting ratio of shRNA-A2AR-injected DG was significantly different from shRNA-Control-injected DG in 4 out of 10 animals (Fig. 4f left graph). An overall analysis of all the animals that developed SE revealed that in shRNA-Control-injected DG, the Δsprouting ratio was not different from zero, showing no impact of shRNA-Control in synaptoporin puncta density. In contrast, shRNA-A2AR-injected DG displayed a significantly negative Δsprouting ratio (P < 0.0001 vs. hypothetical value of 0) and statistically different from the Δsprouting ratio of shRNA-Control (P = 0.0001; paired t-test). These data show that the knockdown of A2ARs in granule cells attenuates synaptoporin puncta density in the inner molecular layer and granule cell layer, demonstrating that A2ARs located in dentate granule cells contribute to SE-induced MF sprouting.
Impact of the pharmacological blockade of A2ARs in SE-induced cell proliferation
MF sprouting into the inner molecular layer occurs in the absence of neurogenesis37. Yet, seizures induce the generation of new dentate granule cells from the subgranular zone38,39,40 and these post-SE born cells also contribute further to MF sprouting, in particular at later stages41,42. Indeed, cells born after SE display hilar basal dendrites and ectopic migration, but do not send axons into the inner molecular layer until 4 weeks after the insult 41,42 and only have a substantial contribution to MF sprouting 10 weeks after SE41.
A2ARs may promote adult neurogenesis particularly in pathological conditions43,44. Hence, we sought to determine if, in addition to the contribution of A2ARs located in dentate granule cells born before SE, A2ARs may also further promote hippocampal MF sprouting by contributing to the generation of newborn cells. To address this, we tested whether the daily administration of the selective A2AR antagonist SCH58261 (0.1 mg/kg, i.p.), beginning 10 h after the onset of SE, affected SE-induced cell proliferation in the granule cell layer. To label mitotically active cells, rats were injected with BrdU at 24 h (200 mg/kg, i.p.), 7 days (200 mg/kg, i.p.) and 8 days (100 mg/kg, i.p.) after SE (Fig. 5a), the latter two time points being within the period of significantly increased mitosis37. Consistent with previous reports38,45,46, quantitative analysis revealed a significant increase in the number of BrdU+-cells in the granule cell layer in SE rats treated with vehicle for 42 days in comparison with control rats (P < 0.001; Fig. 5b,c). The vast majority of BrdU+-cells were co-localized with the mature neuronal marker NeuN (Fig. 5b), in agreement with previous reports describing that the majority (~ 90%) of the newly generated cells within the granule cell layer after SE differentiate into neurons38,45,46. In control rats, SCH58261 administration per se did not significantly modify the density of BrdU+-cells in the granule cell layer, showing that A2ARs do not contribute to basal adult cell proliferation originated from the subgranular zone. Regarding the impact of SCH58261 on SE-induced increase in BrdU+-cells in the DG, we found an interaction between SE and SCH58261 (P = 0.0428; two-way ANOVA). Yet, post-hoc analysis showed that SE-vehicle and SE-SCH58261 groups were not significantly different (P = 0.1492; Fig. 5c). On the other hand, in rats daily injected with SCH58261, SE did not induce a statistically significant increase in BrdU+-cells (P = 0.9722 for SE-SCH58261 vs. Control-SCH58261).
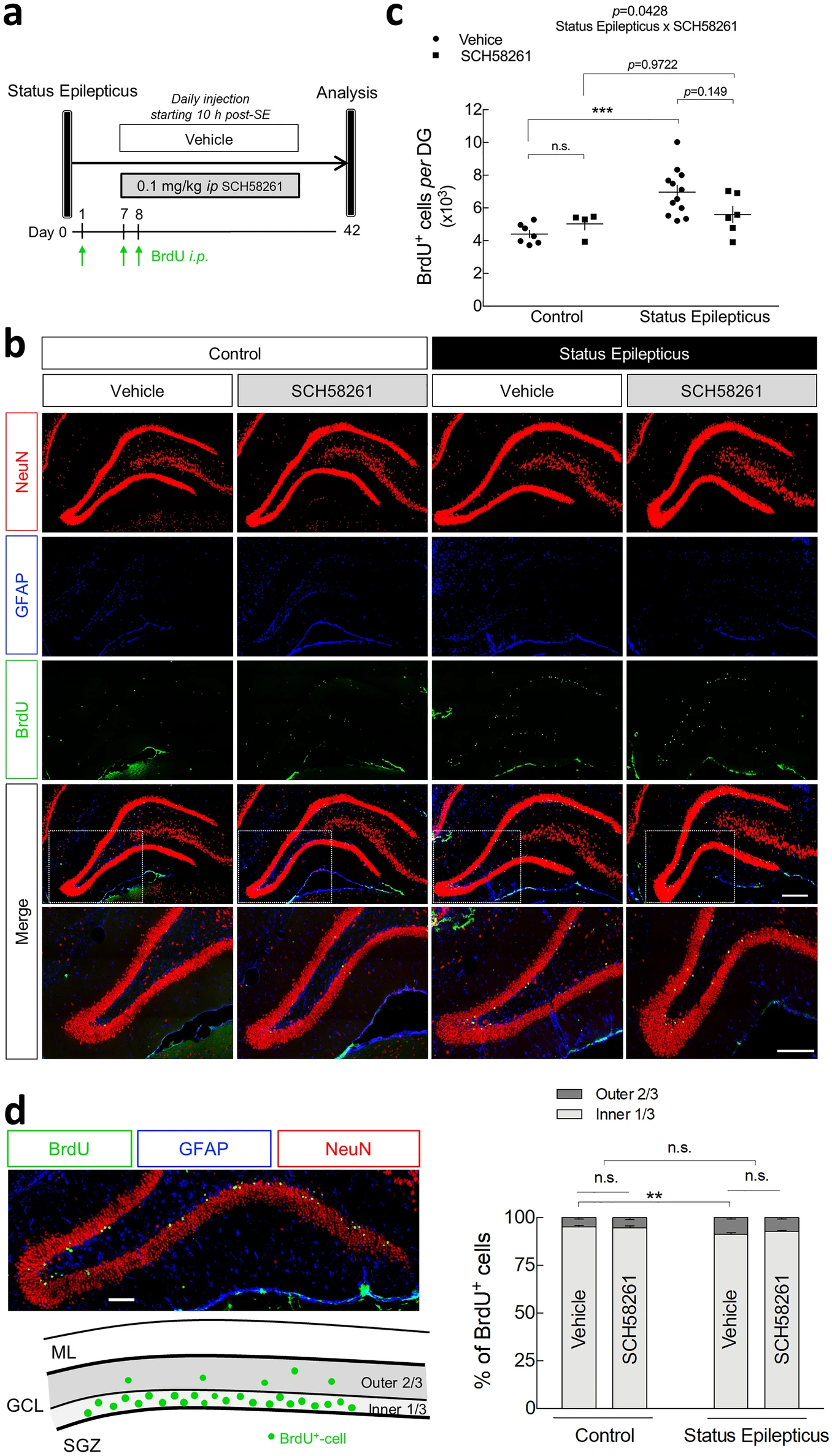
Impact of the pharmacological blockade of A2ARs in status epilepticus-induced cell proliferation and migration of adult-born neurons. (a) Six/seven weeks old rats were injected either with saline (Control group) or with pilocarpine (Status Epilepticus group), 30 min after the administration of scopolamine methylbromide (2 mg/kg, i.p.). Only animals reaching at least stage 4 seizures were considered. Convulsions were suppressed with diazepam (10 mg/kg, i.p.) 2 h after the occurrence of the first stage 4 seizure. The rats from each group were assigned into two subgroups: one subgroup was daily injected with the selective antagonist of A2AR, SCH58261 (0.1 mg/kg i.p.), and the other with vehicle (0.2% Tween-20 in saline), starting 10 h after the onset of status epilepticus. All the animals were i.p. injected with BrdU at 24 h (200 mg/kg), 7 days (200 mg/kg) and 8 days (100 mg/kg) after status epilepticus. BrdU+-cells in the granule cell layer (GCL) per dentate gyrus (DG) was evaluated at 42 days post-status epilepticus by (b) immunohistochemical analysis of BrdU (green), NeuN (magenta) and GFAP (cyan). Scale bar, 300 μm. (c) Status epilepticus induced a significant increase in BrdU+-cells in the GCL per DG in vehicle-injected rats. SCH58261 significantly modified status epilepticus-induced increase in BrdU+-cells density (*P < 0.05 status epilepticus x SCH58261 interaction; two-way ANOVA). Post-hoc analysis revealed that status epilepticus did not significantly increase BrdU+-cells density in rats administered with SCH58261, but also there was no significant difference between status epilepticus-vehicle and status epilepticus-SCH58261. (d) Vehicle-injected rats that experienced status epilepticus displayed a higher relative percentage of BrdU+-cells in the outer two-thirds of the GCL in comparison with the Control group. No interaction was found between SCH58261 and status epilepticus factors (two-way ANOVA). The data are mean ± SEM of the number of the BrdU+-cells in GCL or the relative percentage of cells in the inner (1/3) and in the outer (2/3) parts of GCL. Control-Vehicle, n = 7; Control-SCH58261, n = 4; Status Epilepticus-Vehicle, n = 12; Status Epilepticus-SCH58261, n = 6. ML—molecular layer; GCL—granule cell layer; SGZ—subgranular zone. *P < 0.05, **P < 0.01 and ***P < 0.001, two-way ANOVA with Sidak’s test.
The newborn neurons generated in the subgranular zone migrate radially into the granule cell layer, mainly integrating functionally into the existing circuitry in the inner third of the granule cell layer47,48. Following an epileptogenic insult, there is a misplacement of a significant number of cells that migrate abnormally to the outer two-thirds of granule cell layer38,40. Accordingly, we now observed in control rats that most of the BrdU+-cells were located in the inner one-third of the granule cell layer and only a small portion of BrdU+-cells were located in the outer two-thirds of the granule cell layer (4.82 ± 0.73%; n = 7) (Fig. 5d). SCH58261 administration per se did not modify this pattern in control rats. However, SE rats treated with vehicle for 42 days displayed a significant increase in the relative percentage of BrdU+-cells in the outer two-thirds of the granule cell layer (8.66 ± 0.77%, n = 12), compared to controls (Fig. 5d). In contrast to the observed in SE-induced increase in BrdU+-cells density (Fig. 5c), SCH58261 administration did not modify the SE-induced spatial distribution of adult newborn cells in the granule cell layer (P = 0.3058 for SE x SCH58261 interaction; two-way ANOVA) (Fig. 5d). These data show that A2ARs are not involved in SE-induced aberrant migration of adult-born cells within the granule cell layer.
Status epilepticus-induced hippocampal mossy fiber sprouting is engaged by extracellular ATP-derived adenosine
ATP is preferentially released at high-frequency stimulations and A2ARs are mainly activated by ATP-derived adenosine through the catabolism of extracellular ATP involving ecto-5’-nucleotidase (CD73) activity in MF terminals17,18,19. CD73, which catabolizes extracellular AMP into adenosine, is present in sprouted MF in epileptic rats20 and a SE-induced increased activity of CD73 has been reported in the pilocarpine rat model of temporal lobe epilepsy49,50. CD73 density is also enhanced in the inner molecular layer of the DG from human temporal lobe epilepsy patients51. We now measured a sustained increase of the evoked release of ATP from hippocampal nerve terminals prepared from rats 7 days after pilocarpine-induced SE in comparison with control rats, but not at 24 h or 3 days post-SE (Fig. 6a). This is coincident with the increased CD73 activity found in hippocampal nerve terminals from the same rat model of temporal lobe epilepsy51. This prompted the hypothesis that the adenosine triggering the observed A2AR’s contribution to SE-induced hippocampal MF sprouting derives from the extracellular catabolism of ATP in a CD73-dependent manner. To test this hypothesis, SE-induced MF sprouting was evaluated in CD73-KO mice. The latency to SE onset and the seizure score during SE were similar between wild-type and CD73-KO mice (Fig. 6b). In wild-type mice, SE induced a visible MF sprouting evaluated 105 days later (Fig. 6c). However, in CD73-KO mice, SE induced a significantly lower percentage of the granule cell layer plus molecular layer displaying Timm staining (P = 0.0129 for SE-wild-type vs. SE-CD73-KO), which was not statistically different from control (P = 0.1383 for SE-CD73-KO vs. Control-CD73-KO) (Fig. 6d). This significantly lower SE-induced MF sprouting in CD73-KO mice was also observed along the entire septotemporal axis (Fig. 6e), as observed with A2AR blockade.
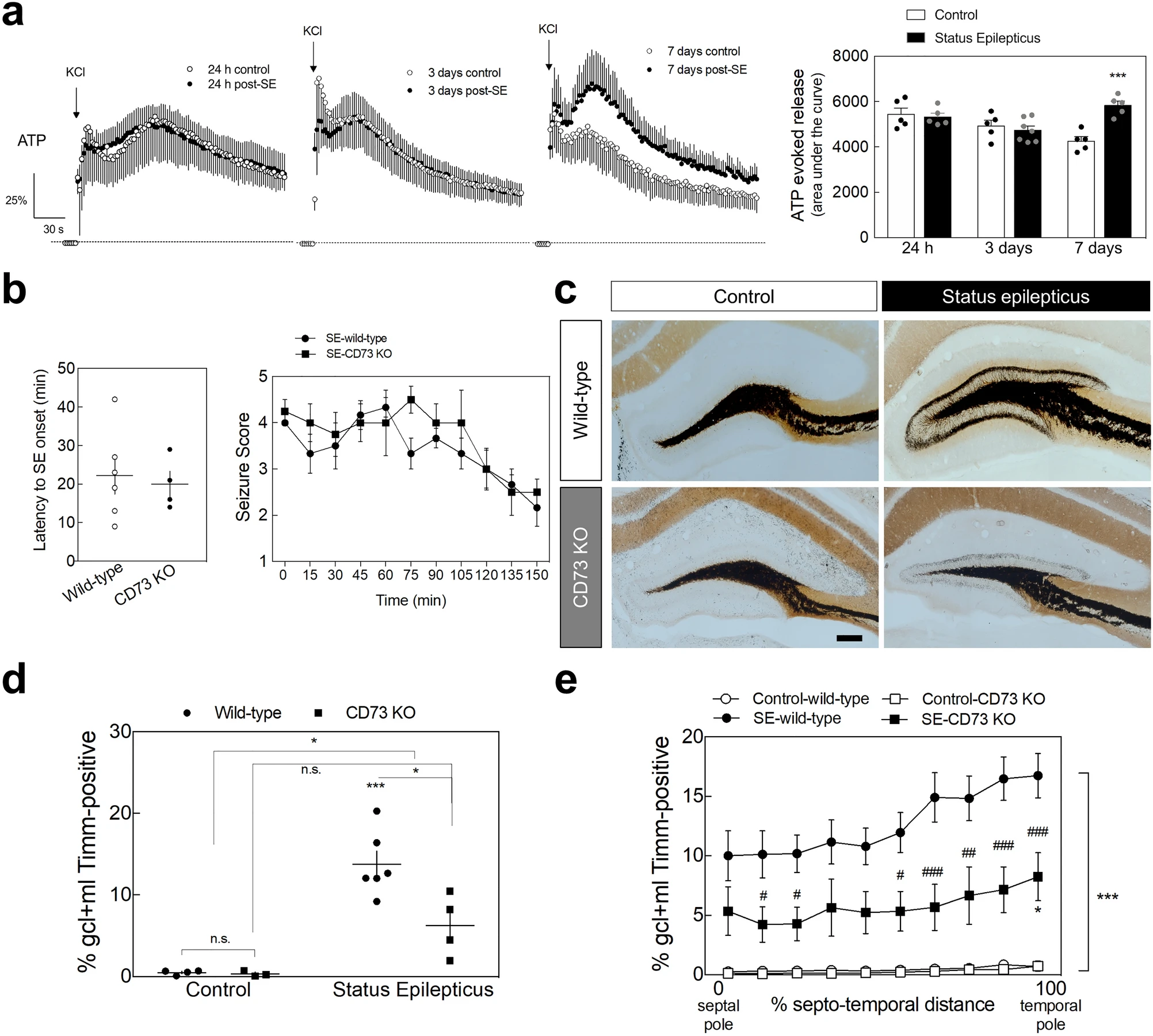
ATP-derived adenosine contributes to status epilepticus-induced hippocampal mossy fiber sprouting. (a) The evoked presynaptic release of ATP induced by a K+-induced depolarization (KCl 30 mM) of purified hippocampal nerve terminals prepared from animals that experienced status epilepticus (SE) was analyzed at 24 h, 3 days and 7 days upon SE induction. Only animals reaching at least stage 4 seizures were considered. Convulsions were suppressed with diazepam (10 mg/kg, i.p.) 2 h after the occurrence of the first stage 4 seizure. While at 24 h and 3 days after SE induction, it was not observed any significant differences, the evoked release of ATP from hippocampal synaptosomes prepared 7 days after SE was significantly higher vs. control rats. Data are mean ± SEM of the percentage of modification in extracellular levels of ATP (time-course graphs) or of the area under the curve (histogram). Control (24 h), n = 5; SE (24 h), n = 5; Control (3 days), n = 5; SE (3 days), n = 7; Control (7 days), n = 5; SE (7 days), n = 5. The data are mean ± SEM of the percentage of modification in extracellular levels of ATP relative to basal levels. ***P < 0.001, one-way ANOVA with Sidak’s test. (b) Latency onset and seizure score during SE of wild-type mice and CD73-KO mice. Data are mean ± SEM. (c) Wild-type mice that had experienced SE displayed a dense Timm staining in the inner molecular layer of the dentate gyrus 105 days later, whereas in CD73-KO mice SE induced a significantly lower Timm staining, as depicted in the representative images (scale bar, 300µm) and quantitatively summarized in the (d) upper histogram. (e) The lower SE-induced mossy fiber sprouting in CD73-KO mice was observed along the entire septotemporal axis of the DG. The data are mean ± SEM of the percentage of the granule cell layer (GCL) plus molecular layer (ML) that displayed Timm staining. Control-wild-type, n = 4; Control-CD73 KO, n = 3; SE-wild-type, n = 6; SE-CD73 KO, n = 4. *P < 0.05 and ***P < 0.001 for SE vs. Control either in wild-type or in CD73-KO mice; #P < 0.05, ##P < 0.01, ###P < 0.001 for CD73-KO vs. wild-type mice either experiencing SE or not; two-way ANOVA with Sidak’s test.
Altogether, these results indicate that the adenosine triggering A2AR-mediated MF sprouting induced by SE is derived from the CD73-mediated extracellular catabolism of ATP.
Discussion
The present study demonstrates that the overactivation of A2ARs is a molecular mechanism underlying synaptic remodeling in the injured brain, namely SE-induced hippocampal MF sprouting. While A2ARs did not contribute to the generation of recurrent excitatory inputs spontaneously developed in dentate granule cells of hippocampal organotypic slices (Fig. 2), the pharmacological blockade of A2ARs significantly attenuated SE-induced hippocampal MF sprouting in a rat pilocarpine model (Fig. 3). This involves A2ARs located in dentate granule cells since the knockdown of A2ARs selectively in dentate granule cells reduced the density of SE-induced MF terminals in the inner molecular layer (Fig. 4). Moreover, the observation of an SE-induced increase in ATP release from hippocampal terminals, together with the prevention/attenuation of hippocampal MF sprouting in CD73-KO mice (Fig. 6), strongly suggests that A2AR-driven MF sprouting is triggered by extracellular ATP-derived adenosine.
We recently reported that A2ARs control neuronal polarization and axon formation of cortical projection neurons during embryogenesis19. In the present study, we found that A2ARs also control axon formation in developing rat hippocampal neurons (Fig. 1). However, in hippocampal neurons, A2ARs did not control the formation of the “normal” axon; instead, the pharmacological activation of A2ARs induced the formation of aberrant secondary axons (Fig. 1). Moreover, we now demonstrated the involvement of A2ARs in SE-induced hippocampal MF sprouting (Fig. 3). This contribution of A2ARs to MF sprouting was only observed in pathological conditions, i.e. in SE-induced MF sprouting, but not in the recurrent excitatory inputs that develop spontaneously in hippocampal organotypic slices (Fig. 2). This indicates that spontaneous MF recurrent sprouting in hippocampal organotypic slices is mechanistically distinct from epilepsy-related hippocampal MF sprouting and argues that an increased activity of A2ARs after SE23 is required to trigger the abnormal axonal sprouting after SE. The observation that the knockdown of A2ARs in granule cells was sufficient to reduce MF sprouting (Fig. 4), together with the ability of A2ARs’ activation to induce the formation of abnormal secondary axons in cultured rat hippocampal neurons (Fig. 1) strongly suggests that A2ARs contribute to hippocampal MF sprouting by directly promoting axon formation/outgrowth selectively during pathological conditions in the adult brain.
This newly described A2AR-mediated mechanism seems intrinsic to neurons rather than involving astrogliosis and inflammation, which are also characteristic features of epilepsy52,53that are associated with the development of seizures54 and with the ability to contribute to synaptic remodeling55. Although both astrogliosis and neuroinflammation are also controlled by A2ARs17, it was previously reported that blocking A2ARs abrogates SE-induced neurodegeneration without affecting astrogliosis24 and it was now observed that the selective elimination of A2ARs in hippocampal neurons was sufficient to prevent SE-induced MF sprouting. Thus, the proposed conclusion that A2AR-mediated control of SE-induced MF sprouting seems to involve an intrinsic neuronal remodeling by the adenosine neuromodulation system, seems the most parsimonious explanation, which may entail a direct regulation of cytoskeleton, as observed in developing rat cortical neurons56. Nevertheless, MF sprouting has been also shown to be correlated with cell loss, although it is not mandatory57. In particular, the extension of hippocampal MF sprouting has been correlated with the loss of hilar inhibitory interneurons and mossy cells57 found both in experimental models and in patients with temporal lobe epilepsy58,59,60,61,62,63. Another study also reported a positive correlation between MF sprouting and cell loss both CA3 and CA164. As mentioned, the antagonism of A2ARs prevents SE-induced neurodegeneration23. This has been associated to an up-regulation of neuronal/synaptic A2AR-driven increase in excitatory drive leading to calpain-mediated neurodegeneration23. More recently, it was observed an absence of neurodegeneration upon SE in CD73-KO mice24. The involvement of A2ARs and of CD73 in hilar neurons death was not detailed23,24. Nevertheless, there is the possibility of an additional indirect contribution of the CD73-A2AR axis to hippocampal MF sprouting by controlling hippocampal cell damage.
This study determined that the contribution of A2ARs to SE-induced hippocampal MF sprouting here identified entails, at least in part, A2ARs located in dentate granule cells generated before SE (Fig. 4). But newly born cells also contribute to the abnormal retrodirective innervation of the inner molecular layer38,41,42. There are evidences suggesting for an inhibitory action of A2ARs in neurogenesis in postnatal and adult rodent brain, in particular in cell proliferation44. A2ARs were shown to contribute to the transient decrease in cell proliferation from the subgranular zone upon oxygen–glucose deprivation, most likely by contributing to the damage of immature cells21. On the other hand, it was recently reported that the activation of A2ARs prevents the reduction of newborn dentate granule cells as a consequence of hearing loss induced by noise exposure43. Here, we observed that the pharmacological blockade of A2AR per se did not alter adult-born cell generation from the subgranular zone (Fig. 5). However, we found a statistically significant interaction between SCH58261 and SE in cell proliferation, but not in their positioning within the granule cell layer (Fig. 5). This suggests that SE-induced dentate granule cell proliferation, but not their migration, entails, either directly or indirectly, the activation of A2ARs. Hence, A2ARs may further contribute and sustain MF sprouting by promoting the generation of adult born cells induced by epileptogenic injury. The newborn cells only contribute to the abnormal innervation of the inner molecular layer upon their full maturation within 4 weeks41,42, although immature granule cells are able to extend axons to CA365,66. Thus, it will be now interesting to elucidate if A2ARs contribute to MF formation/outgrowth independently of the postsynaptic target or if A2ARs are only involved in the growth of abnormal retrodirective axons. In addition, it is worth noting that we recently showed that A2ARs also play a key role in GABAergic synapses stabilization20. Although we cannot yet extend this role of A2ARs to the stabilization of MF synapses, we do not discard also an eventual postsynaptic effect of A2ARs in the stabilization/formation of MF contacts. Hence, our findings leave open the possibility that A2ARs may control circuit remodeling in the adult brain not only by the presynaptic regulation of axogenesis, either directly or indirectly, but also through the reactivation of their ability to control synapse stabilization/maturation.
The gain of function of neuronal A2ARs after SE to control MF sprouting provides a functional correlate between the previously reported increased density of A2ARs in nerve terminals after SE22 and the increased density of CD73, which has been proposed to be a marker of synaptic remodeling after SE51. The present observations that the synaptic release of ATP is increased after SE (Fig. 6a) and that the knockout of CD73 dampens SE-induced MF sprouting (Fig. 6b–e) allows concluding that noxious brain conditions such as SE trigger an upregulation of the whole ATP-CD73-A2AR axis to control MF sprouting. Although this bolstered ATP-CD73-A2AR axis seems to be recurrently engaged in the control of brain and synaptic dysfunction in different animal models of brain diseases24,67,68, the molecular mechanisms engaged by A2ARs to tinker with synaptic remodeling are still not defined. The control of synaptogenesis during development involves an A2AR-mediated control of cAMP levels in synaptic terminals20, but the A2AR-mediated control of synaptotoxicity after noxious stimuli seems instead to involve p38-mediated signaling69. A possible A2AR-mediated control of BDNF and TrkB function70 is also grounded on conflicting evidence: although several evidences point towards an involvement of an up-regulation of BDNF in triggering this initial branching9,10,11,13,71, this does not seem sufficient per se to induce MF sprouting12,14. Furthermore, both the A2AR-mediated control of synaptogenesis during development as well as the A2AR-driven axonal elongation in cultured cortical neurons were not modified by the presence of a BDNF scavenger20,56. Clearly, the transducing pathways operated by these pleiotropic A2ARs to control MF sprouting still remains to be unraveled.
Since the selected model to induce a pathological-like sprouting process in the adult brain is also relevant to model epileptogenesis, it is of interest to tentatively frame the present findings in the context of epilepsy. Adenosine has been for long known as an anti-convulsant72 via A1 receptors (A1R) activation73, which are mostly activated by adenosine released per se, depressing basal excitatory transmission74. However, during epileptogenesis there is a down-regulation of A1Rs indicated by both a decreased density of A1Rs in excitatory synapses in animal models of epilepsy22 and a reduction of ambient levels of adenosine, mostly driven by an increased activity of adenosine kinase75,76. In contrast, A2ARs become prominent at high-frequencies stimulations, activated by adenosine originated from the CD73-dependent extracellular catabolism of ATP that is prominently released at high-frequencies17,18,24. Moreover, it was reported an increased density of A2ARs in different animal models of epilepsy22,77,78 and in patients with epilepsy79, together with the prominent synaptic release of ATP at seizure-like firing patterns17,80,81,82 and an increase of the density and activity of CD7324,49,50,51,79, which supports a gain of function of A2ARs during epileptogenesis. Indeed, as mentioned, A2ARs tether SE-induced increase in neuronal excitability to hippocampal neurodegeneration23, an effect that is dependent on CD73 24. Moreover, although we have not directed the present study to investigate this question, it is interesting to note that several studies demonstrated anti-convulsant effects upon inhibition of A2ARs83,84,85,86. More importantly, the ability of the pharmacological inhibition of A2ARs to mostly prevent the progressive seizure severity in kindling protocols23,25,86 and to arrest long-term deleterious consequences of early-life convulsions87, strongly indicate a relevant contribution of A2ARs to epileptogenesis. This is in accordance with the presently concluded ability of A2ARs to contribute to the circuit remodeling characteristic of epileptogenesis. However, despite the evidence indicating that MF sprouting forms recurrent excitatory inputs3,4,88,89,90,91,92, the link between MF sprouting and progressive hyperexcitability and seizure severity is still a working hypothesis. It may be pro-epileptogenic, homeostatic or an epiphenomenon2,93 or may be contributing to epilepsy comorbidities rather than seizure severity94. Interestingly, A2ARs and CD73 also contribute to some of the comorbidities associated to epilepsy23,24. Further studies are now needed to unravel the functional consequences of the involvement of A2ARs and CD73 in epilepsy-related circuit remodeling here identified.
Overall, the findings presented shown that A2ARs contribute to epilepsy-related circuit remodeling, which may involve the reactivation of the ability of A2ARs to control axon formation and outgrowth during development19. This posits A2ARs as potential targets to manipulate aberrant synaptic remodeling during epileptogenesis, which may be extended to other disorders displaying also circuit alterations such as neuropsychiatric disorders affecting adults and prevalent on ageing.