
Abundant Aβ fibrils in ultracentrifugal supernatants of aqueous extracts from Alzheimer’s disease brains

By Andrew M. Stern,Yang Yang,Shanxue Jin,Keitaro Yamashita,Angela L. Meunier,Wen Liu,Yuqi Cai,Maria Ericsson,Lei Liu,Michel Goedert,Sjors H.W. Scheres, and Dennis J. Selkoe
Excerpt from the article published in Neuron, May 10, 2023 DOI: https://doi.org/10.1016/j.neuron.2023.04.007
Highlights
- Aβ in aqueous brain extracts (oligomers, protofibrils) are insoluble and fibrillar
- The diffusible Aβ fibrils have the same atomic structure as amyloid plaque fibrils
- Lecanemab binds to and protects against the synaptotoxicity of diffusible fibrils
Summary
Soluble oligomers of amyloid β-protein (Aβ) have been defined as aggregates in supernatants following ultracentrifugation of aqueous extracts from Alzheimer’s disease (AD) brains and are believed to be upstream initiators of synaptic dysfunction, but little is known about their structures. We now report the unexpected presence of Aβ fibrils in synaptotoxic high-speed supernatants from AD brains extracted by soaking in an aqueous buffer. The fibrils did not appear to form during preparation, and their counts by EM correlated with Aβ ELISA quantification. Cryo-EM structures of aqueous Aβ fibrils were identical to those from sarkosyl-insoluble homogenates. The fibrils in aqueous extracts were labeled by lecanemab, an Aβ aggregate-directed antibody reported to improve AD cognitive outcomes. Lecanemab provided protection against aqueous fibril synaptotoxicity. We conclude that fibrils are abundant in aqueous extracts from AD brains and have the same structures as those from plaques. These findings have implications for AD pathogenesis and drug design.
Introduction
Amyloid β (Aβ) and tau monomers aggregate into fibrils found in amyloid plaques and neurofibrillary tangles in the brains of people with Alzheimer’s disease (AD). Fibrils can be extracted from postmortem brains by ultracentrifugation in the presence of detergents such as N-lauryl sarcosine (sarkosyl). Pelleted detergent-insoluble fibrillar Aβ, however, has been reported as less toxic than Aβ from aqueous ultracentrifugal supernatants prepared without detergent.1 Aqueous supernatants contain Aβ aggregates that disrupt synaptic plasticity1,2,3 and neurite morphology,4,5 promote tau phosphorylation,4,6,7 decrease memory,1 and accelerate Aβ seeding.8,9,10,11 These aqueous supernatant Aβ aggregates have been assumed to be non-fibrillar and termed “soluble oligomers” or “protofibrils.” Their neurotoxicity makes them therapeutic targets.
Lecanemab is an FDA-approved monoclonal antibody that improved cognitive outcomes in a phase 3 trial.12 Its success has been attributed to selectivity for soluble Aβ protofibrils over insoluble fibrils.13,14 However, a structural definition of protofibrils (or oligomers) from human brains does not exist.15 Here, we use “aggregate” instead of protofibril or oligomer because oligo implies few monomers, whereas aggregates in aqueous brain extracts may comprise thousands of monomers. We use “protofilament” (not protofibril, which has no strict definition) to mean a single stack of β-pleated sheet-folded monomers—for Aβ, two protofilaments form one fibril.16 Nomenclature notwithstanding, improving AD therapy requires a structural understanding of aqueous Aβ aggregates in the human brain.
The term “soluble” is used for aggregates found in aqueous supernatants, but ultracentrifugation protocols vary and are often poorly described. Most Aβ aggregates aqueously extracted from AD brains have been reported to be high molecular weight (HMW): they elute in or near the void volume of size exclusion chromatography (SEC) columns (≥500 kDa).17,18 Low molecular weight (LMW) aggregates from AD brain, including covalent dimers,19 are particularly toxic,1,18 but disassembly of HMW aggregates ex vivo is often necessary to observe LMW aggregates and their toxicity.1,4,18 The structure of any of these is unknown.
Dynamic non-covalent interactions render extraction of aqueous Aβ aggregates under non-denaturing conditions challenging. A recent method soaks minced bits of AD cerebral cortex instead of homogenizing tissue, to avoid shearing plaques and other large Aβ aggregates—these soaking extracts exhibited a similar toxicity to homogenates.20,21 We previously reported a non-denaturing method to enrich for Aβ aggregates from AD soaking extracts by immunoprecipitation with the calcium-sensitive antibody B24 and were surprised to observe Aβ fibrils in the B24 eluates of putatively soluble extracts.22 We now report that Aβ fibrils are present in aqueous soaking extracts even without immunoprecipitation, have the same structure as plaque fibrils, inhibit synaptic transmission, and are bound and blocked by lecanemab.
Results
Aβ fibrils can be pelleted from aqueous extracts of AD brain
For immunoprecipitation using B24,22 we started with aqueous extracts prepared by mincing and soaking multiple regions of AD cerebral cortex in Tris-buffered saline (TBS), followed by ultracentrifugation and collection of the supernatant (Table 1).20 We previously immunoprecipitated Aβ with B24 from these soaking extracts in the presence of calcium and eluted it using ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), to avoid conditions which might denature aggregates.22 We found that the eluted HMW Aβ aggregates could be re-pelleted in a benchtop centrifuge at 20,000 g in a 1.5-mL tube for 1 h (pelleting distance ∼1 cm), with no detectable Aβ remaining in the supernatants by ELISA. Immunogold labeling showed short Aβ fibrils in the pellets.22
| Step # | Steps in standard protocol | Modifications of standard protocol for the specified control experiments, and relevant figure number |
|---|---|---|
| 1 | Frozen tissue thawed, gray matter dissected | Figures S3I–S3L: fresh tissue |
| 2 | Tissue chopped using McIlwain tissue chopper set to 0.5 mm width | – |
| 3 | Tissue bits soaked in 1:5 w:v extraction buffer 30 min 4°C with nutation in a 50-mL tube | Figure S2B: tissue bits first rinsed with ∼15 volumes extraction buffer before soaking Figure S3E: 1:15 w:v |
| 4 | 2,000 g fixed angle (pelleting distance ∼4 cm) 10 min 4°C, top 90% supernatant collected | – |
| 5 | 200,000 g in SW41Ti (pelleting distance ∼9 cm) 110 min 4°C, top 90% of supernatant collected | Figure S2A: top 90% of supernatant divided into top 2 mL and bottom ∼7 mL Figures 1C–1G: 20,000–475,000 g in TLA100.3 (pelleting distance ∼2 cm) for 110 min 4°C; top 90% of supernatant collected |
| 6 | 1 mL aliquotted into 1.5-mL Eppendorf LoBind tubes, stored at −80°C | Figure S2E: aliquotted into siliconized or BSA-coated tubes Figures S3C and S3H: stored at 4°C Figure S3D: diluted serially before freezing Figures S3E and S3F: added various Aβ aggregation inhibitors before freezing Figure S3G: filtered through aluminum oxide filters before freezing |
| 7 (re-centrifugation) | Aliquot(s) thawed, 20,000 g fixed angle (pelleting distance ∼1 cm) 60 min 4°C. Input, supernatant and pellet retained for ELISA and/or EM | – |
The soaking extract method and its modifications for control experiments in this report
We now asked if Aβ fibrils could be pelleted from soaking extracts without immunoprecipitation. We spun 1 mL of TBS soaking extracts (Table 1, step #7) at 20,000 g for 1 h. Soluble Aβ aggregates would not be expected to pellet. However, a substantial proportion of aggregated Aβ (i.e., that which was detectable by a monomer-preferring Aβ ELISA only after disassembly in 5 M GuHCl) was depleted from the supernatants and recovered in the pellets (Figure 1A, center). Monomeric Aβ (i.e., not requiring GuHCl denaturation for detection by the monomer-preferring ELISA) was not depleted by re-centrifugation (Figure 1A, right), as expected. Examining the re-centrifugation pellets by EM revealed amyloid fibrils that reacted with N-terminal anti-Aβ antibody D54D2 (Figure 1B). In aqueous extracts from the minced AD brains, we also noticed fibrils resembling paired helical filaments (PHFs) that stained with anti-tau antibodies (Figure S1). In this report, we focus on the Aβ fibrils. None were observed in the pellets from aqueous extracts of two control brains (C1 and C2), and a single fibril was seen on an EM grid of a third control brain (C3). The Aβ fibrils’ average length across thirteen brains was ∼100 nm (Figure S2A). However, a quantile-quantile plot (Q-Q plot) suggested undercounting of both very short fibrils (due to an inability to see them) and very long fibrils (due to their exceeding the micrograph size) (Figure S2B). Thus, there was probably a wider distribution of lengths than depicted in Figure S2A.
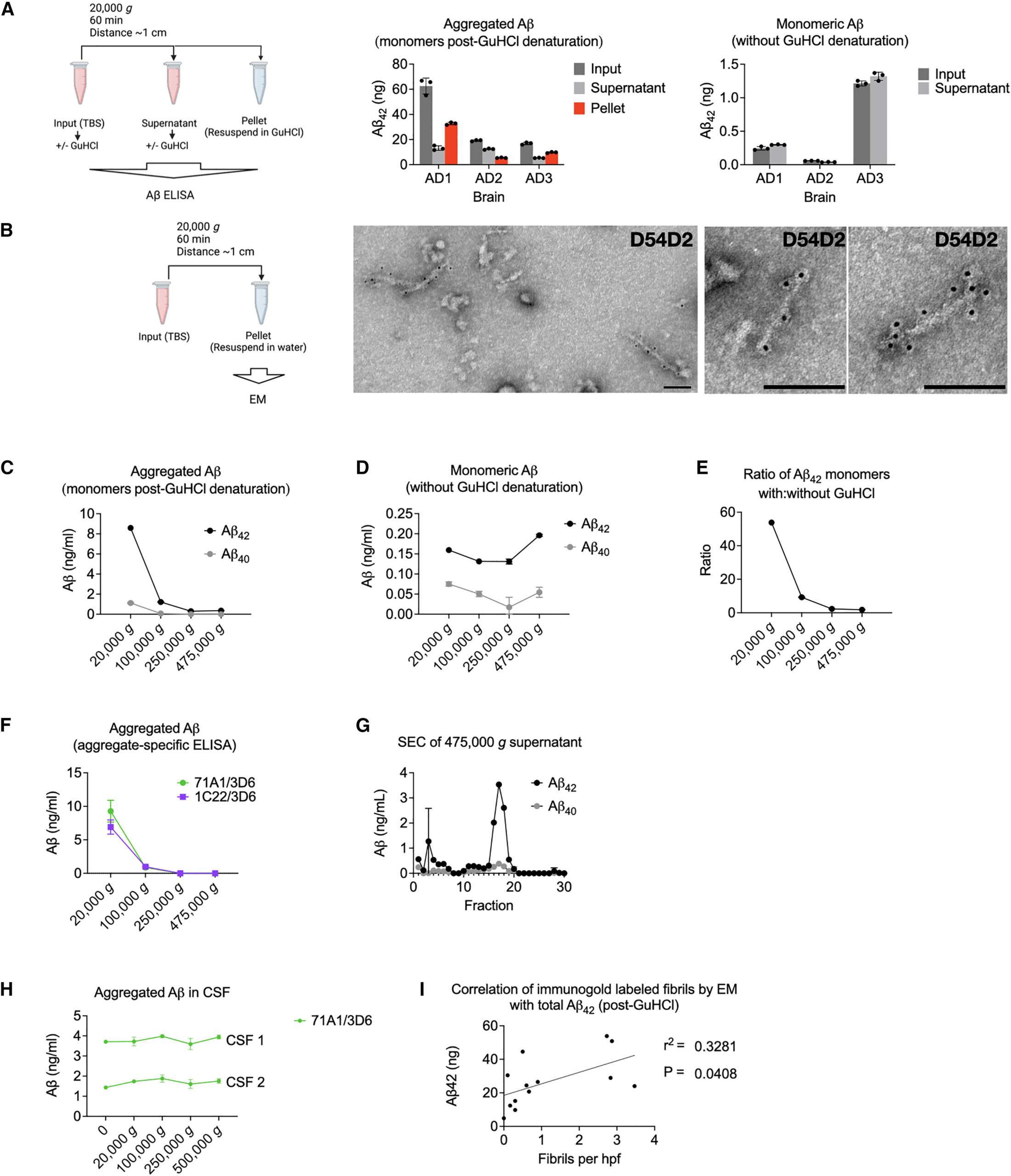
Aqueous soaking extracts of AD brain contain insoluble Aβ fibrils
Aβ fibrils were not contaminants from the original ultracentrifugation pellets because they were present when only the top ∼20% of the supernatant was collected and re-pelleted (Figure S2C). To rule out that the fibrils had adhered to the surface of the minced tissue rather than diffused out of the tissue into the soaking buffer, we rinsed the minced brain bits with 15 volumes of extraction buffer through a 100-μm filter prior to soaking. We found no reduction in the amount of extracted Aβ aggregates measured biochemically (Figure S2D) or as fibrils seen by EM. Further arguing in favor of diffusion of fibrils from the minced tissue into the aqueous buffer rather than carryover from the cut surface was the observation (also in Figure S2 of Hong et al.20) that diffusion of Aβ into the buffer required 15–30 min to plateau, similar to the diffusion rate of control soluble proteins.
Sufficient centrifugal force can pellet HMW Aβ aggregates
When accounting for the pelleting distance, our initial and re-centrifugation protocols were approximately equal in efficiency: the distance in thin-wall tubes in the SW41Ti rotor (initial soaking extract) is ∼9 cm when full (Table 1, step #5), whereas it is ∼1 cm for a 1 mL microcentrifuge tube at a 45° angle in the benchtop centrifuge (Table 1, step #7). Thus, the benchtop centrifugation should accomplish the same pelleting efficiency at an ∼9-fold lower g-force. It was therefore possible that if higher g-forces had been used initially in preparing the soaking extracts, the fibrils would not be recovered from the supernatant during re-centrifugation. To test this, we modified the initial ultracentrifugation, using the fixed-angle TLA100.3 rotor (pelleting distance ∼2 cm) at increasing g-forces, instead of the SW41Ti rotor (pelleting distance ∼9 cm). We found that g-forces ≥250,000 g across the shorter distance depleted nearly all biochemically detectable aggregated Aβ from the supernatants (Figure 1C) but retained native monomers (Figure 1D). In 475,000 g supernatants, GuHCl denaturation released only 1.8-fold more Aβ monomers than without GuHCl, whereas in 20,000 g supernatants, GuHCl addition released 54-fold more (Figure 1E). This difference suggests that most Aβ in the 475,000 g supernatants consisted of native monomers and covalent dimers.1,19
To verify that Aβ aggregates were depleted from supernatants spun ≥250,000 g, we used two high-sensitivity ELISAs with aggregate-preferring capture antibodies23,24 not requiring GuHCl denaturation (Figure 1F). Neither assay could detect aggregates in the ≥250,000 gsupernatants. Residual Aβ in the 475,000 g supernatants exhibited an LMW SEC profile (Figure 1G).
We conclude that most Aβ, and all its HMW aggregates from aqueous brain extracts, is made of suspended particles, consistent with the presence of fibrils. We compared this finding with Aβ in cerebrospinal fluid (CSF). Detection of CSF Aβ by aggregate-specific ELISA was low and unaffected by centrifugal force (Figure 1H). This suggests that the few Aβ aggregates detected in CSF were soluble, in contrast to those from aqueous extracts of AD brain.
Aβ fibrils in aqueous soaking extracts do not appear to form during sample preparation
We investigated if the Aβ fibrils were absent during life but polymerized from smaller non-fibrillar aggregates during the postmortem interval or sample processing. We used the standard soaking extract protocol using the SW41Ti rotor at 200,000 g,20 followed by re-centrifugation in the tabletop centrifuge at 20,000 g (Table 1).20
One possibility was that Aβ polymerization occurred along the plastic tube surface during re-centrifugation of the original soaking extracts. We re-centrifuged the standard soaking extracts and moved the supernatants to new tubes at specific time intervals. The Aβ in the pellets left behind was quantified by ELISA following GuHCl denaturation. If new fibril nucleation were occurring, we would have expected a lag in the accumulation of Aβ in the pellets, whereas pelleting of pre-existing particulates would be linearly time-dependent until they had been depleted from the supernatants. We observed no lag, suggesting no nucleation (Figures S2E and S2F). Storage of soaking extracts in siliconized or bovine serum albumin-coated tubes had no effect on subsequent re-pelleting compared with standard Eppendorf Protein LoBind polypropylene tubes (Figure S2G).
We investigated if Aβ polymerization occurred during the freezing and thawing of freshly prepared soaking extracts. When proceeding directly from the initial SW41Ti rotor (Table 1, step #5) to a second spin in the benchtop centrifuge (step #7) at 4°C, Aβ aggregates measured by ELISA could still be pelleted (Figure S3A), and fibrils were observed by immuno-EM (Figure S3B). Nevertheless, freezing and thawing of a freshly prepared soaking extract resulted in the re-pelleting of more Aβ compared with storage at 4°C (Figure S3C). We also found that the re-pelleting was concentration-dependent: when the original ultracentrifugation supernatants were diluted at least 4-fold before freezing, less aggregated Aβ could subsequently be re-pelleted after thawing (Figure S3D). This concentration dependence only occurred when dilution was performed after soaking and initial centrifugation. When dilution was performed by increasing the tissue weight-per-volume (w:v) soaking ratio from the usual 1:5–1:15 g/mL, similar amounts of Aβ were extracted, and the freeze/thaw dependence of the amount pelleted remained (Figure S3E). These results suggest that at w:v ratios of 1:5 and 1:15, the extraction of Aβ saturated at similar concentrations.
To assess further whether polymerization occurred during freeze/thaw, we added Aβ aggregation inhibitors, including aggregate-binding antibodies, 0.45% CHAPS,25 and scyllo-inositol,26,27 immediately after ultracentrifugation of the soaking extracts and before freezing the supernatants. None of these agents reduced the amount of Aβ that could be re-pelleted after thawing (Figures S3F and S3G). We also expected that if fibrils had formed ex vivo from monomers and/or soluble LMW aggregates during freeze/thaw, filtering the soaking extracts immediately after ultracentrifugation should have had no effect on the recovery and pelleting of Aβ. We found that freshly prepared soaking extracts were depleted of Aβ aggregates by a 20-nm pore size alumina filter (Figure S3H) but that they passed through a 100-nm filter, implying that Aβ aggregates were at least 20 nm in size prior to freezing the original soaking extracts. SEC of a freshly prepared TBS extract immediately after ultracentrifugation (without freezing) showed that Aβ still eluted in the void volume of a Superdex 200 Increase column (∼≥500 kDa) (Figure S3I). Together, these results suggest that Aβ aggregates were HMW prior to freezing and storage of the soaking extracts. Increased pelleting of Aβ fibrils after freeze/thaw may be due to the clumping of existing fibrils but is probably not due to new fibril formation.
Another possibility was that polymerization occurred after death but before tissue freezing. To best exclude this artifact, we prepared an AD soaking extract and applied it to the Superdex 200 Increase column within 8 h of death without freezing either the tissue or the extract. Virtually all the ELISA-detectable aggregated Aβ eluted in the void volume of a Superdex 200 Increase column (Figures S3J–S3L), and fibrils could be pelleted upon re-centrifugation (Figure S3M).
Fibrils account for a substantial proportion of HMW Aβ aggregates in soaking extracts
We counted the D54D2-immunoreactive fibrils in the re-centrifugation pellets from 13 AD brains, using 30 EM fields each. The average number of fibrils per field correlated with the Aβ levels by ELISA after GuHCl denaturation (Figure 1I). The 13 brains appeared to separate into two groups, but our limited sample size could not determine if this was a chance separation or two true populations.
Fibrils from soaking extracts of AD brain have the same structures as those from sarkosyl-insoluble fractions
The cryoelectron microscopy (cryo-EM) structures of Aβ fibrils from sarkosyl-insoluble homogenates of AD brains were recently determined.16
In comparison, the aqueous soaking extract-derived pellets showed shorter fibrils that clumped less, consistent with their relative resistance to pelleting. Cryo-EM structure determination of aqueous soaking extract-derived fibrils from two cases of sporadic AD (AD7 and AD11) revealed identical structures to those of Aβ fibrils from sarkosyl-insoluble homogenates 18 (Figure 2). AD7 had only type I fibrils, whereas AD11 had type I and type II fibrils.16
Tau fibrils from the re-centrifuged aqueous soaking extracts were structurally identical to PHFs from sarkosyl-insoluble homgenates of AD brains (Figure S4).28
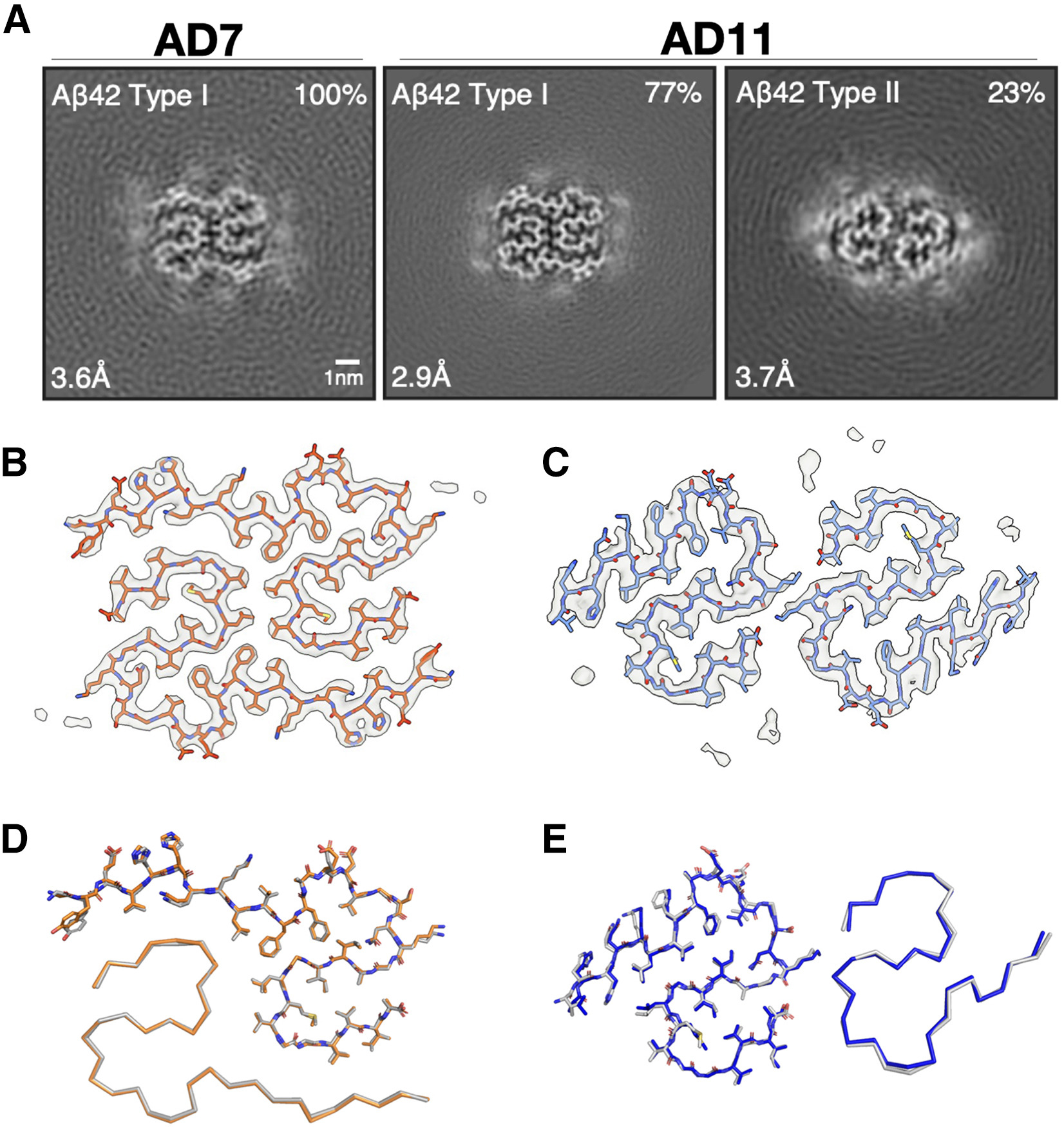
Aβ fibrils from aqueous extracts of AD brains have the same cryo-EM structures as fibrils from sarkosyl-insoluble homogenates
Lecanemab labels Aβ fibrils from soaking extracts and protects from synaptotoxicity
Lecanemab is a humanized mouse monoclonal antibody raised against synthetic aggregates of Aβ bearing the AD-causing amyloid precursor protein (APP) “Arctic” mutation E693G.13,29
Synthetic Aβ40[E693G] forms more soluble aggregates than wild-type Aβ40 in vitro, and the mechanism of action of lecanemab is thought to relate to its preference for small, soluble Aβ aggregates over fibrils.14
Since lecanemab has been shown to immunoprecipitate Aβ from aqueous AD brain extracts,13 we asked whether it can bind to fibrils. Lecanemab decorated Aβ fibrils (Figures 3A and 3B) that had been re-pelleted from AD aqueous soaking extracts. We also examined antibody h1C22, which we previously reported binds better to synthetic Aβ oligomers than monomers or mature fibrils.5,22,23
We observed specific h1C22 labeling of Aβ fibrils from aqueous saoking extracts (Figures 3C–3E).
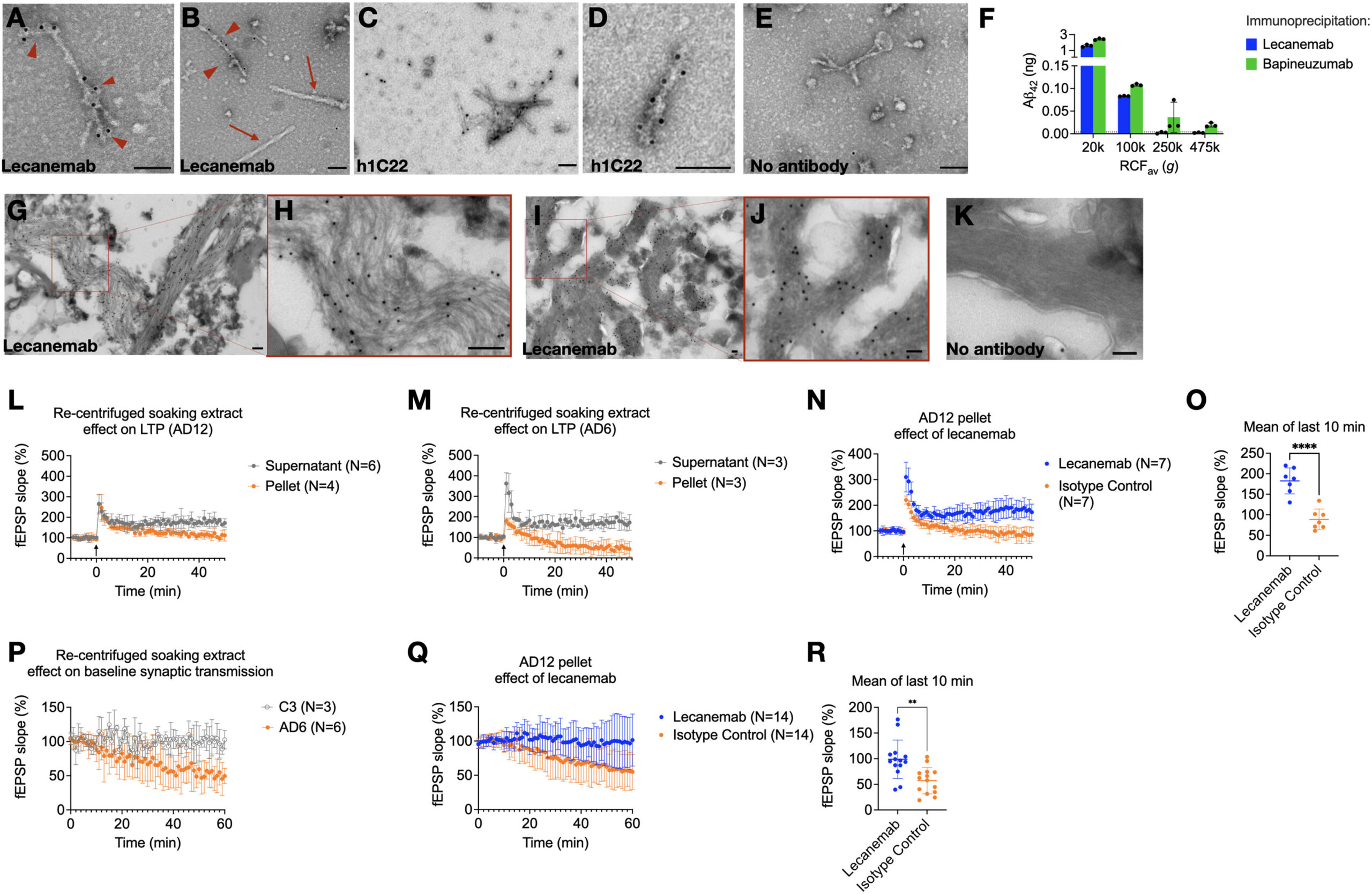
Lecanemab decorates Aβ fibrils from Alzheimer’s disease brains and protects from synaptotoxicity
Since lecanemab labeled Aβ fibrils from aqueous brain extracts, we hypothesized that it would only immunoprecipitate Aβ from extracts prepared at g-forces that retain aggregates detectable by other means, such as aggregate-preferring ELISAs.24
We found that lecanemab could only immunoprecipitate Aβ from aqueous AD brain supernatants that had been ultracentrifuged at ≤100,000 g in the TLA100.3 rotor (Figure 3F), like 1C22 (Figure 1F). Bapineuzumab, which binds both monomeric and aggregated Aβ, could still immunoprecipitate some Aβ from 250,000 and 475,000 g supernatants (Figure 3F).
Lecanemab stains plaques and vascular amyloid by immunohistochemistry.30
To determine if it stains plaque fibrils, we prepared minimally fixed ultrathin cryosections from AD cortex to preserve antibody epitopes. Although cytoarchitecture was not well preserved, lecanemab labeled fibrils in situ(Figures 3G–3J), with little background from protein A gold alone (Figure 3K).
We previously found that aqueous extracts of AD brain, but not control brain, can impair long-term potentiation (LTP) in wild-type mouse hippocampal slices.1,18,22,31
We asked if synaptotoxicity in this model was in the insoluble or soluble fraction of aqueously diffusible soaking extracts. Re-centrifugation demonstrated synaptotoxicity in pellets but not supernatants (Figures 3L and 3M). Lecanemab reversed synaptotoxicity by a mean of 93.7% field excitatory post-synaptic potential (fEPSP) slope compared with isotype control (95% CI, 60.6%–127%, p < 0.0001) (Figures 3N and 3O). We observed that the resuspended fibril-rich pellets could impair the baseline field potential slope without LTP induction by high-frequency stimulation (HFS) (Figure 3P). Pellets from a re-centrifuged control brain soaking extract (C3) did not impair baseline synaptic transmission (Figure 3P). Lecanemab prevented inhibition of the synaptic baseline compared with isotype control (mean difference 42% fEPSP slope, 95% CI 17%–67%, p = 0.002) (Figures 3Q and 3R).
Discussion
Our results indicate that at least some Aβ aggregates from aqueous extracts of AD brains are identical in structure to plaque-derived fibrils. Although we cannot rule out the presence of some non-fibrillar HMW Aβ aggregates, a correlation between total Aβ levels and the number of Aβ fibrils suggests that fibrils are a substantial proportion of HMW Aβ aggregates (oligomers and protofibrils) that diffuse from AD cerebral cortex into aqueous extracts.
An unavoidable limitation of this work is the need to examine AD brains postmortem. Although we attempted to mitigate artifacts, we cannot exclude the disassembly of some plaques into free-floating fibrils after death. We also cannot exclude some ex vivoaggregation of monomers or LMW aggregates (e.g., dimers) into fibrils. However, small-pore filters, inhibitors of Aβ aggregation, and the use of fresh tissue provided no evidence for these artifacts.
If short, dispersed Aβ fibrils can diffuse through the brain, they may be in equilibrium with plaques. APP transgenic mice show halos surrounding methoxy-XO4-labeled plaques that immunoreact with Aβ aggregate-preferring antibodies.32
These halos may consist of Aβ fibrils that are packed too loosely to shift the fluorescence spectra of thioflavin dyes and have more access to synaptic endings than fibrils from plaque cores. Supporting this model is our observation that diffusible Aβ fibrils have the same cryo-EM structures as those from sarkosyl-insoluble brain fractions. Experiments in mice in which microglia were depleted, or the AD risk gene TREM2 was knocked out, have shown that microglial plaque compaction may protect dendrites and synapses.33,34,35
Our observation of synaptotoxicity within the fibril-containing fractions of soaking extracts is compatible with this model.
Lecanemab prefers intermediate molecular weight synthetic Aβ aggregates over synthetic fibrils13; however, in humans, lecanemab reduces amyloid positron emission tomography (PET) signal, a marker of fibrillar Aβ,12,36 and labels amyloid plaques and blood vessel deposits by immunohistochemistry.30
Based on our finding that lecanemab labels Aβ fibrils in aqueous AD brain extracts, and that both lecanemab binding and lecanemab-dependent synaptotoxicity were restricted to the pelletable fractions, we conclude that lecanemab’s therapeutic target in the human brain includes diffusible fibrils with the same structures as those from plaques. It remains possible that lecanemab’s target includes non-fibrillar aggregates because we could not exclude their presence.
We also observed PHFs in soaking extracts. Others have observed that tau seeding activity in human aqueous extracts depends on the largest molecular weight tau aggregates.10,11
Based on our results, we suspect that these seed-competent preparations contained PHFs with identical folds to those from sarkosyl-insoluble preparations.37
Supporting this model is the observation that short tau fibrils were the most seed-competent species in brain extracts from transgenic mice expressing human P301S tau.38
Future design of therapeutic agents against tau seeds should consider that they may be PHF.