
A011, a novel small-molecule ligand of σ2 receptor, potently suppresses breast cancer progression via endoplasmic reticulum stress and autophagy
By Yuyun Li, Xiaoyang Xie, Shiyi Liao, Zhanwei Zeng, Siyan Li, Baocheng Xie, Qunfa Huang, Huan Zhou, Chenhui Zhou, Jiantao Lin, Yunsheng Huang, and Daohua Xu
Excerpt from the article published in Biomedicine & Pharmacotherapy, Volume 152, 2022, 113232, DOI:
https://doi.org/10.1016/j.biopha.2022.113232.
Editor’s Highlights
- The σ2 receptor ligand compound A011 had no measurable affinity for σ1 receptor (Ki > 1000 nM), but had a high affinity for σ2 receptor (Ki = 77.1 ± 2.6 nM).
- The σ2 ligand significantly inhibited the proliferation of various cancer cells in a concentration-time-dependent manner.
- The σ2 ligand had better inhibitory effect on MCF-7, MDA-MB-231 and A549 cells than cisplatin, and had less toxic effect on normal human mammary epithelial MCF-10A cells than cisplatin.
- The σ2 ligand induces G0/G1 and G2/M phase cell cycle arrest and apoptosis
- The σ2 ligand induced the increase of intracellular ROS and Ca2+concentration
- The σ2 ligand induced the increase of autophagy in MCF-7 cells
- The σ2 ligand inhibited the activation of the PI3K-Akt-mTOR pathway and activated the PERK-eIF2α-CHOP pathway
- The σ2 ligand induced autophagy and apoptosis of tumor cells but showed no obvious toxicity toward main organs in vivo
Authors’ Highlights
- A011 had high affinity and selectivity for σ2 receptor.
- A011 induced caspase-dependent apoptosis in MCF-7 cells.
- A011 induced endoplasmic reticulum stress in MCF-7 cells.
- A011 showed obvious anti-tumor activity and no significant toxicity in an in vivo tumor model.
Abstract
Breast cancer has surpassed lung cancer to become the most commonly diagnosed cancer in women worldwide. Sigma-2 (σ2) receptor is considered to be a potential therapeutic target for breast cancer because of its high expression in breast cancer cells and low expression in normal breast cells. Many σ2 ligands have been reported to have excellent anticancer activity, but their mechanism of action has not been fully elucidated. We discovered that A011 had high affinity and selectivity for σ2receptor, reduced proliferation in five cancer cell lines, and significantly inhibited the monoclonal formation ability of MCF-7 cells. Furthermore, A011 rapidly increased the levels of intracellular Ca2+ and reactive oxygen species and induced autophagy. Molecular pharmacology studies revealed that A011 induced endoplasmic reticulum stress, activated the PERK-eIF2α-CHOP pathway and inhibited the activation of the PI3K-Akt-mTOR pathway, leading to cell apoptosis. In an in vivo tumor model, A011 showed obvious anti-tumor activity and no significant toxicity. More importantly, our study demonstrated for the first time that endoplasmic reticulum stress is the main mechanism of anti-cancer effects for σ2ligands, at least for A011. A011 may potentially be useful as a therapeutic agent for treating breast cancer.
Graphical Abstract
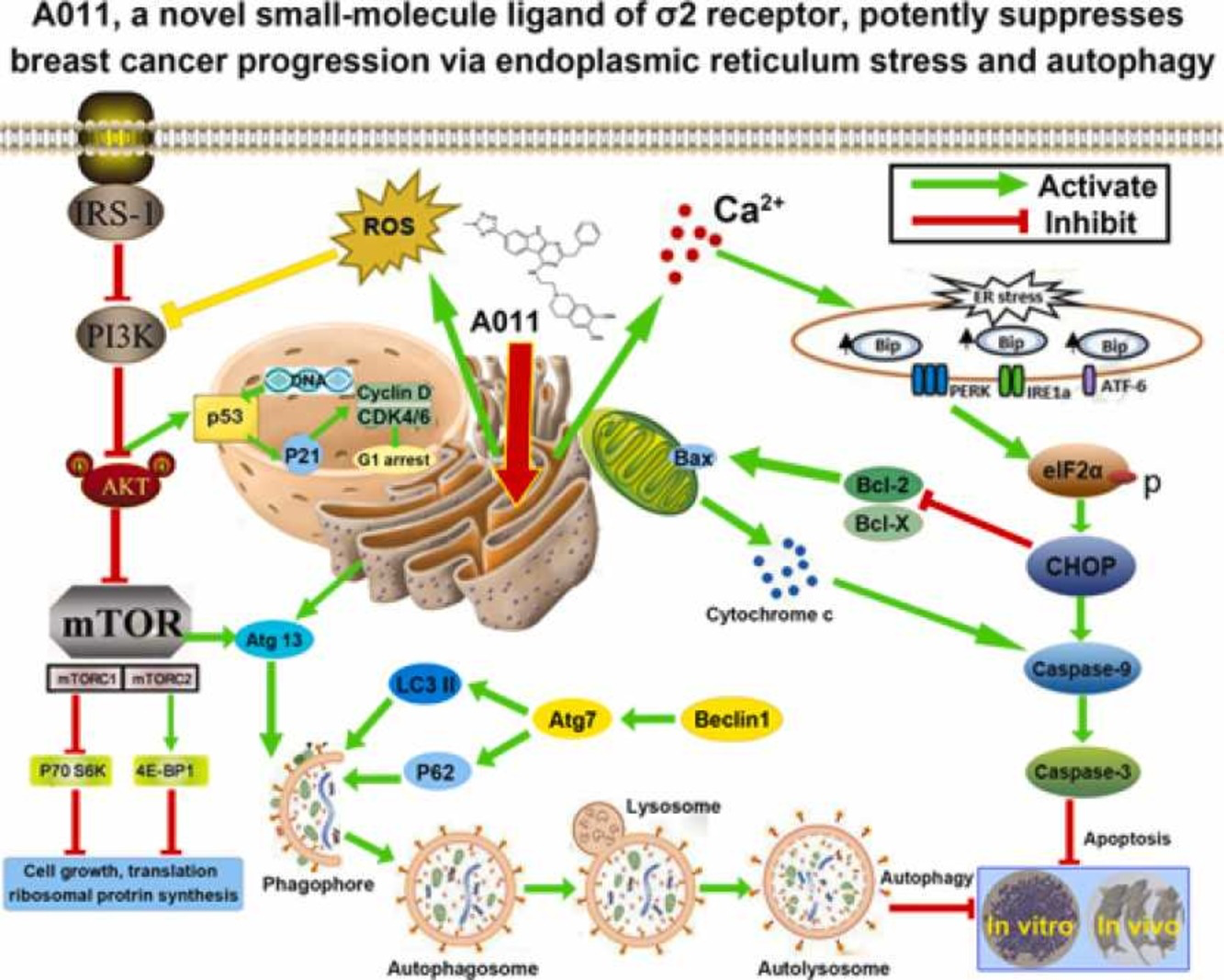
1. Introduction
According to the International Agency for Research on Cancer, breast cancer has surpassed lung cancer to become the most commonly diagnosed cancer worldwide by 2020 [1]. Although overall mortality for breast cancer patients has declined, it remains the most common cause of death from cancer in women [2], [3]. In recent years, various therapies emerged in the area of breast cancer, including surgery, radiotherapy, neoadjuvant and adjuvant therapy [4]. However, side effects, multidrug resistance and metastasis are still the main obstacles to achieve an effective treatment [5], [6]. Therefore, clarifying the mechanism of invasion, metastasis and drug resistance of breast cancer and exploring new therapeutic targets are necessary for the development of targeted drugs and new diagnostic and therapeutic protocols, which are of great significance for improving the therapeutic outcome for breast cancer patients.
The σ receptors are a class of proteins that were once mistakenly described as a subtype of the opioid receptors [7]. Subsequent studies revealed that they were distinguished intracellular proteins and there were two subtypes, named σ1 and σ2[8]. The σ1 receptor protein has been elucidated and was cloned in 1996 [9], whereas the σ2 receptor was isolated by Alon and coworkers and confirmed to be identical to TMEM97, an endoplasmic reticulum (ER) resident transmembrane protein [10], [11]. The crystal structure of σ2 complex and its exact physiological function in cells are still to be identified. A growing interest in σ2 receptor is largely due to its involvement in diseases such as cancer and neurological disorders [12], [13], [14]. Studies have demonstrated that the σ2 receptor densities in proliferating cancer cells are almost 10 folds higher than those in quiescent cancer cells and thus σ2receptor is regarded as a potential biomarker of the proliferative status of solid tumors [15], [16]. Various σ2 ligands have been reported to have strong inhibitory effects against various cancer cells and no or low toxicity to normal cells both in vitroand in animal models [16], [17]. Pharmacological studies have indicated that σ2ligands induce the production of reactive oxygen species (ROS), stimulate intracellular Ca2+ release from its storage, block cell cycle, promote autophagy and induce caspase-dependent and caspase-independent apoptosis in cancer cells [18], [19], [20]. Despite of the progresses, the common mechanism by which σ2 ligands exert their anticancer effects and the inner associations between them are still unclear.
The endoplasmic reticulum (ER) is a complex and dynamic organelle involved in many cellular functions, including production and folding of cellular proteins, calcium homeostasis, carbohydrates and lipids metabolism [21]. The realization of ER function depends on a series of equilibrium factors in its internal environment, such as glycosylinvertase activity, calcium ion level and redox potential. When these equilibrium factors are out of balance, misfolded proteins will accumulate in the ER lumen, disturb ER homeostasis and eventually lead to ER stress [22]. Cells have an integrated signaling system to respond to the ER stress and restore ER homeostasis. Fundamental pathways mainly include the unfolded protein response (UPR), ER-associated degradation and autophagy. Among them, autophagy plays a dual role in regulating cells’ fates: mild autophagy protects cells from harmful conditions and promotes cell survival; while severe or rapid autophagy will induce cell apoptosis [23]. The synergistic activity of all these processes determines the extent of ER stress and thus governs whether ER will reestablish homeostasis or activate cell death programs [24].
σ2 ligands are potentially useful for cancer diagnosis, treatment or adjuvant therapy [25], [26]. However, the exact physiological role of this receptor and its endogenous ligand are still unclear, and the lack of structural information has seriously hindered the elucidations of its physiological and pharmacological roles as well as the development of more selective ligands for σ2 receptor. We discovered that 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline scaffold was a pharmacophore for σ2receptor and that compounds containing this moiety bound to σ2 receptor with high affinity and selectivity [27]. Subsequent studies revealed some analogs containing 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline benzamide structures demonstrated good anticancer activity against common and multidrug resistant tumors [28]. More recently, we synthesized a new series of 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline analogs without the benzamide moiety and found that those analogs had even higher affinity and selectivity for σ2 receptor [29], [30]. Through further structural modification, we identified a potent and selective novel σ2 ligand, A011,which has the 6,7-dimethoxytetrahydroisoquinoline structure and showed high affinity for σ2 receptor and no measurable affinity for σ1 receptor. We further clarified for the first time that A011 induced ER stress, caused the rapid increase of intracellular ROS and Ca2+ levels, promoted autophagy, blocked cell cycle and induced caspase-dependent apoptosis in breast cancer MCF-7 cells. Furthermore, A011 showed obvious anti-tumor activity and no significant toxicity to normal tissues in an in vivo tumor model. Our results suggest that A011 is potentially useful as a therapeutic agent for treating breast cancer.
…
3. Results
3.1. The binding affinities of A011 to σ receptors
The binding affinities of A011 to both σ receptors were evaluated by radioligandbinding analysis. It was found that A011 had no measurable affinity for σ1 receptor (Ki > 1000 nM), but had a high affinity for σ2 receptor (Ki = 77.1 ± 2.6 nM). These data indicated that A011 was a selective σ2 receptor ligand. The chemical structure of A011 is shown in Fig. 1 A. The concentration-affinity curve of A011 competitively binding to σ2 receptor is shown in Fig. 1B.
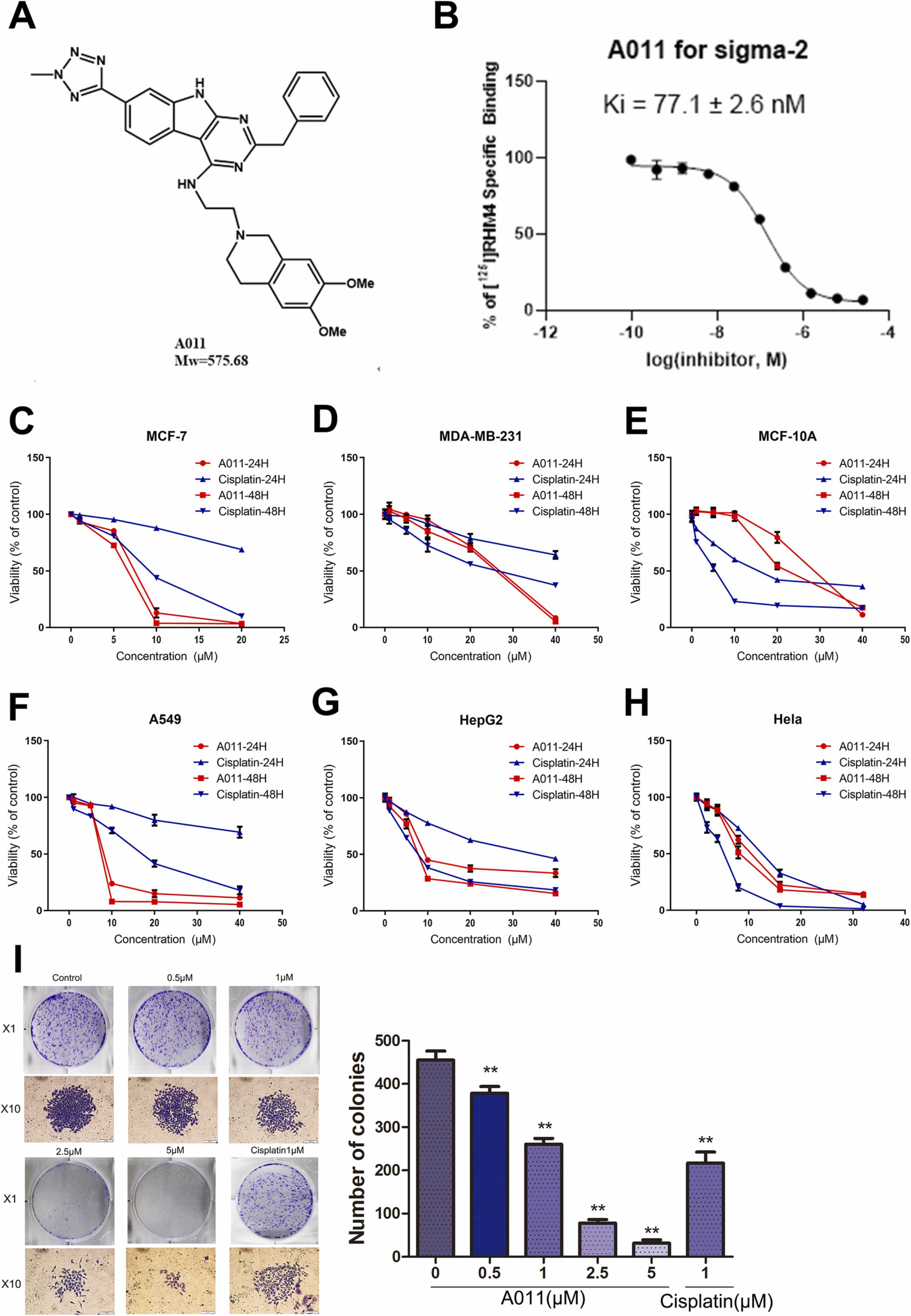
The structure and cytotoxicity of A011. (A) The structure of A011. (B) The concentration-affinity curve of A011 competitively binding to σ2 receptor. (C-H) The cytotoxicity of A011 against MCF-7, MDA-MB-231, MCF-10A, A549, HepG2 and HeLa, respectively. Cells were treated with a range of concentrations of A011 or cisplatin for 24 h and 48 h. (I) The inhibitory effect of A011 against the monoclonal formation ability of MCF-7 cells. Data represent mean ± SD of three different experiments.*P < 0.05, * * P < 0.01 vs Control.
3.2. The cytotoxicity of A011 on various human cancer cell lines
Studies have demonstrated that the expression level of σ2 receptor is abnormally elevated in some types of rapidly proliferating cancer cells, such as breast cancer, non-small cell lung cancer, oral squamous cell carcinoma, colorectal cancer and gastric cancer, especially in poorly differentiated cancer cells but not in normal cells [32]. We examined the cytotoxicity of A011 and cisplatin on five types of human cancer cell lines and normal human mammary epithelial cells by CCK-8 assay. The results showed that A011 significantly inhibited the proliferation of various cancer cells in a concentration-time dependent manner (Fig. 1C-H). Furthermore, A011 had better inhibitory effect on MCF-7, MDA-MB-231 and A549 cells than cisplatin, and had less toxic effect on normal human mammary epithelial MCF-10A cells than cisplatin (Table 1). In addition, A011 significantly inhibited the monoclonal formation ability of MCF-7 cells (Fig. 1I). The above data indicated that A011 had excellent anti-proliferation activity against breast cancer cells. MCF-7 cells have been thoroughly characterized for the σ2 receptor expression and have become a model for σ2-related investigations [33]. Therefore, MCF-7 cells were selected as a representative cancer cell line for subsequent experiments.
| IC50 (μM) | |||||||
|---|---|---|---|---|---|---|---|
| Compounds | MCF-7 | MDA-MB-231 | A549 | HepG2 | HeLa | MCF-10A | |
| A011 | 5.87 ± 0.09 | 19.39 ± 2.17 | 7.13 ± 0.17 | 6.85 ± 0.32 | 8.49 ± 0.09 | 22.42 ± 1.27 | |
| Cisplatin | 8.92 ± 0.15 | 24.53 ± 2.79 | 15.71 ± 1.22 | 5.27 ± 0.18 | 4.10 ± 0.16 | 11.01 ± 0.69 |
3.3. A011 induces G0/G1 and G2/M phase cell cycle arrest and apoptosis
Various σ2 receptor ligands have been reported to inhibit cancer cells growth by disrupting cell cycle and promoting apoptosis [20], [34]. In this study, we investigated the effects of A011 on cell cycle and apoptosis. Flow cytometry assays showed that the proportion of MCF-7 cells in G0/G1 and G2/M phase significantly increased after 12 h treatment with A011 (Fig. 2 A). At the same time, the MCF-7 cells were also observed undergoing apoptosis after 24 h treatment with A011, in which the proportions of apoptotic cells increased from 4.77 ± 0.62% to 62.10 ± 1.59% (Fig. 2D). The qRT-PCR assay was used to detect the expression of cell cycle and apoptosis-related genes in MCF-7 cells after 12 h treatment with A011. The results showed that cell cycle related genes cyclin D1 and cyclin B1 were significantly down-regulated (Fig. 2B). The apoptosis-related genes Bad, Bax and caspase-3 were significantly up-regulated, whereas Bcl-2 was significantly down-regulated (Fig. 2E). Western blot results showed that after 24 h treatment with A011, the expression of p53 protein in MCF-7 cells was significantly increased, while cyclin D1 and cyclin B1 proteins were significantly decreased (Fig. 2 C). The expression of apoptotic-related proteins of Bad and Bax were significantly up-regulated, and Bcl-2 protein was significantly down-regulated. Meanwhile, the expression of cleavage proteins of caspase-3 and PARP-1 were significantly increased (Fig. 2 F). To investigate whether A011 induced apoptosis is caspase dependent, a pan caspase inhibitor FMK was applied. In combination administration, FMK significantly inhibited apoptosis of MCF-7 cells induced by A011 (Fig. 2 G) and decreased the cleavage proteins of caspase-3 (Fig. 2H). These results suggest that A011 induces caspase dependent apoptosis in MCF-7 cells.
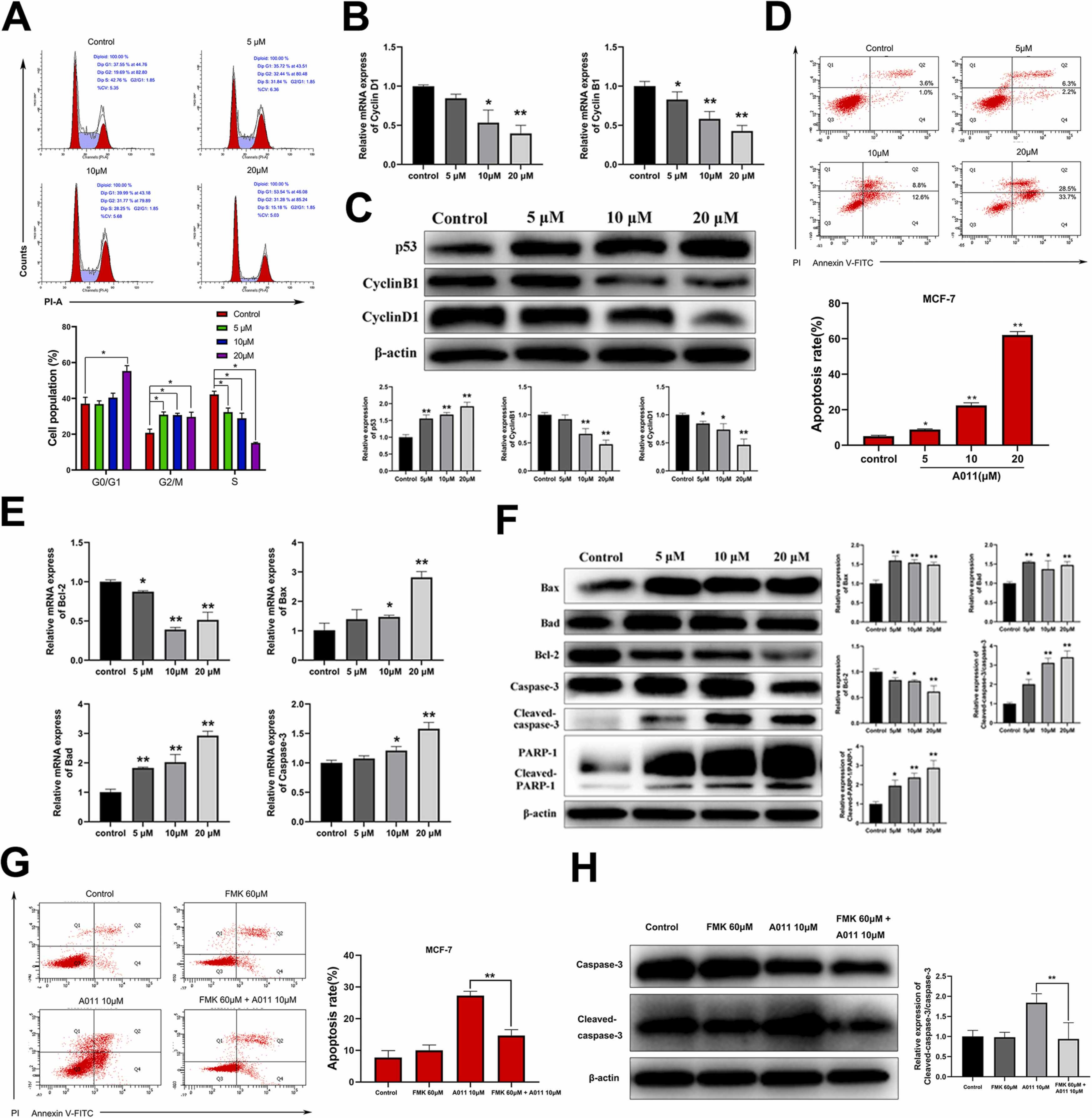
A011 induces cell cycle arrest and apoptosis in MCF-7 cells. (A) A011 induced cell cycle G0/G1 and G2/M phase arrest. Cells were treated with A011 (5, 10, 20 μM) for 12 h and then detected by flow cytometry. (B) A011 induced significant down-regulation of cyclin D1 and cyclin B1 genes. Cells were treated with A011 for 12 h and then detected by qRT-PCR assay. (C) A011 significantly increased the expression of p53 protein, but decreased the expression of cyclin D1 and cyclin B1 proteins. Cells were treated with A011 for 24 h and then detected by western blot assay. (D) A011 induces the apoptosis of MCF-7 cells in vitro. Cells were treated with A011 for 24 h and then detected by flow cytometry. (E) A011 induced significant up-regulation of Bad, Bax and Caspase-3 genes, but down-regulation of Bcl-2 gene. (F) A011 significantly increased the expression of Bad, Bax, Cleaved-caspase-3 and Cleaved-PARP-1 proteins, but decreased the expression of Bcl-2 protein. (G) FMK significantly inhibited apoptosis of MCF-7 cells induced by A011. Cells were treated with A011 in the presence or absence of FMK for 24 h and then detected by flow cytometry. (H) FMK significantly decreased the expression of cleaved-caspase-3 proteins in MCF-7 cells induced by A011. Cells were treated with A011 in the presence or absence of FMK for 24 h and then detected by western blot assay. Data represent mean ± SD of three different experiments. *P < 0.05, * *P < 0.01 vsControl.
3.4. A011 induced the increase of intracellular ROS and Ca2+concentration
An interesting phenomenon is that various σ2 ligands can induce the production of ROS in cancer cells [35], [36], and at the same time induce Ca2+ efflux from the ERand mitochondria [37], [38]. In this study, DCFH-DA probe was used to detect the level of ROS in MCF-7 cells. The experimental results showed that, after A011 intervention for 4 h, the level of intracellular ROS was significantly increased, and the application of vitamin E reduced the elevated ROS level induced by A011 in MCF-7 cells (Fig. 3 A). CCK-8 assay showed that vitamin E could reverse the A011-mediated proliferation toxicity in MCF-7 cells to a certain extent (Fig. 3B). Similarly, Fluo-3/AM probe was used to detect the level of free Ca2+ in MCF-7 cells. The experimental results showed that A011 (5 μM, 10 μM) and siramesine (10 μM) could significantly increase the level of intracellular free Ca2+ after 6 h intervention (Fig. 3 C). When the concentration of A011 increased to 20 μM, the level of intracellular free Ca2+ was significantly increased after 0.5 h of administration (Fig. 3D).
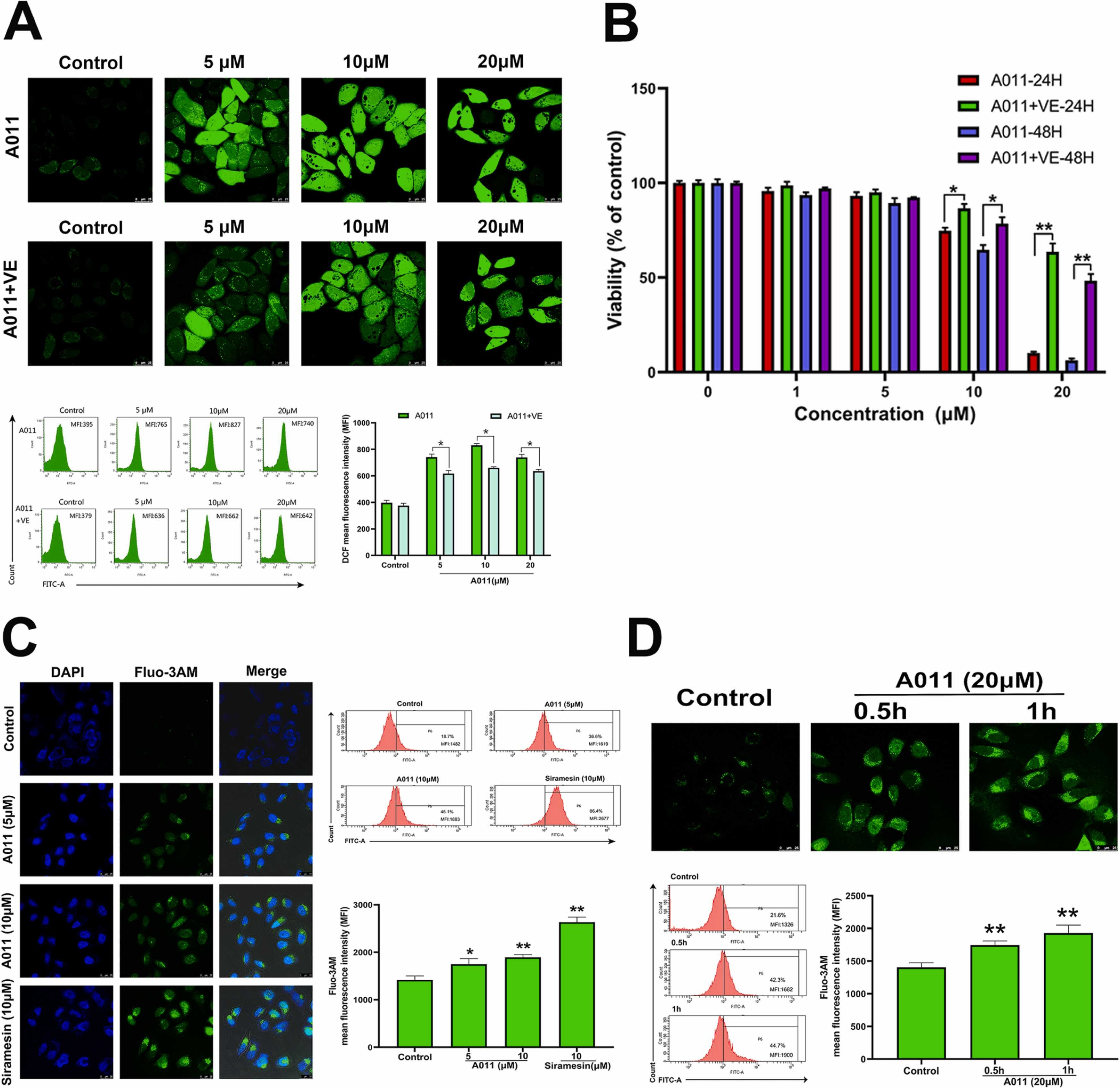
A011 induced the increase of intracellular reactive oxygen species (ROS) and Ca2+concentrations. (A) Effect of A011 alone or in combination with vitamin E on the level of ROS in MCF-7 cells. ROS was detected by laser confocal microscopy and flow cytometry after 4 h intervention of A011 alone or in combination with vitamin E. (B) The reverse effect of vitamin E on A011-mediated proliferation toxicity of MCF-7 cells was detected by CCK-8 assay. (C) The effect of A011 with different concentration on free Ca2+ levels in MCF-7 cells. The free Ca2+ level was detected by laser confocal microscopy and flow cytometry. (D) The effect of A011 with different duration of intervention on free Ca2+ levels in MCF-7 cells. The free Ca2+ level was detected by laser confocal microscopy and flow cytometry. Data represent mean ± SD of three different experiments. *P < 0.05, * *P < 0.01 vs Control. Scale bar = 25 µm.
3.5. A011 induced the increase of autophagy in MCF-7 cells
Autophagy is a lysosomal degradation process that removes misfolded proteins, cytoplasmic components and dysfunctional organelles to maintain cellular homeostasis [22]. A well-orchestrated program including over 30 autophagy-related genes (Atg) regulates autophagy, which can be induced by nutrient starvation and subsequent inhibition of mechanistic target of rapamycin (mTOR) signaling, or by the UPR as misfolded proteins accumulate [39], [40]. Previous studies have shown that some types of σ2 ligands can induce autophagy in cancer cells [41]. In this study, the autophagy flow in MCF-7 cells was detected by Adplus-mCherry-GFP-LC3B adenovirus. Adenovirus infected target cells can effectively express the fusion proteins of red fluorescent protein (mCherry), green fluorescent protein (GFP) and LC3B protein, presenting bright red and green fluorescence. In the case of non-autophagy, the fusion proteins exhibited the combined effect of mCherry and GFP, and thus exhibited diffuse yellow fluorescence in the cytoplasm. In the case of autophagy, the fusion proteins aggregate on the membrane of autophagosome and represented as yellow spots. When autophagosomes fuse with lysosomes, they appear as red spots due to the quenching of GFP fluorescence. Therefore, by observing the color of mCherry-GFP-LC3B fusion protein, the autophagy flow of cells can be indicated. The experimental results showed that MCF-7 cells in the control group presented diffuse yellow fluorescence, while yellow and red spots appeared in cells in the A011 treatment group. With the increase of A011 concentration, the number of spots gradually increased, indicating the increase of autophagosomes and autophagosomes-lysosomes in cells (Fig. 4 A). The observation of autophagy structure under transmission electron microscopy is considered as the gold standard of autophagy. We observed more autophagosomes and autophagosomes-lysosomes in MCF-7 cells after A011 treatment by transmission electron microscopy compared with the control group, suggesting that A011 increased autophagy in MCF-7 cells (Fig. 4B). Western blot results showed that after A011 treatment for 12 h, the expression of p62 protein in MCF-7 cells significantly decreased, and the expression of Beclin 1 protein significantly increased. The LC3-II/I ratio significantly increased (Fig. 4 C). In combination administration, 3-MA, a pan autophagy inhibitor, partially reversed the decrease in MCF-7 cells viabilityinduced by A011 (Fig. 4D). In addition, we found that A011 induced the formation of various vacuoles in MCF-7 cells after 12 h of administration (Fig. 4E), which was also observed in the σ2 ligand WC-26 [20]. In fact, these large vesicles with autophagosome-like structures are referred to ER-containing autophagosomes [42]. UPR in many cells leads to ER expansion, which is accompanied by the formation of ER-containing autophagosomes that are densely and selectively packed with membrane stacks derived from the UPR-expanded ER [43].
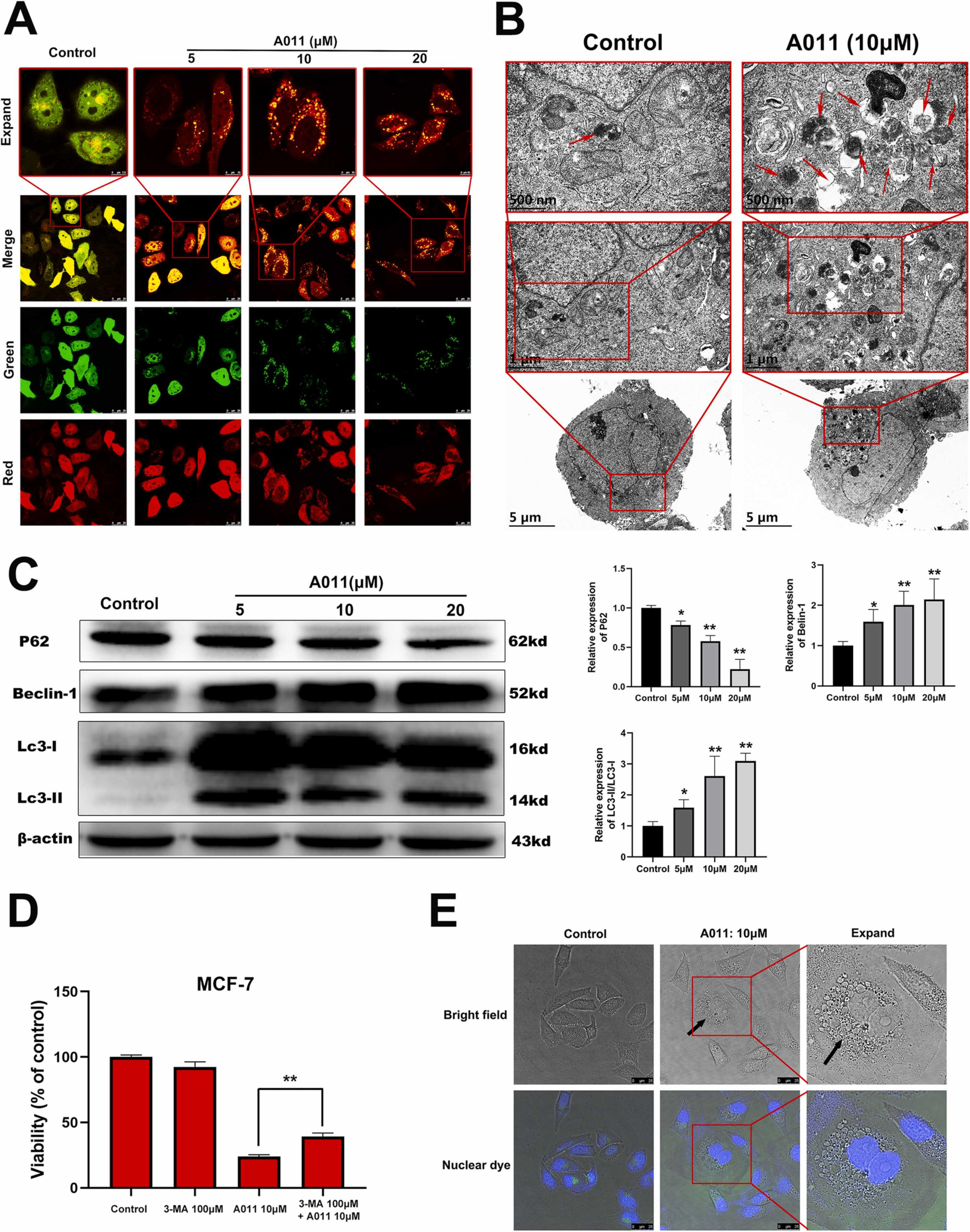
A011 induces the increase of autophagy in MCF-7 cells. (A) The autophagic flow was detected by Adplus-mCherry-GFP-LC3B adenovirus transfection after A011 treatment for 6 h. (B) The autophagosomes was detected by transmission electron microscopy after A011 treatment for 6 h. (C) A011 significantly increased the expression of Beclin 1 and LC3-II protein, while decreased the expression of p62 protein. The protein content of cells treated with A011 for 12 h was detected by western blot assay. (D) 3-MA partially reversed the decrease in MCF-7 cells viability induced by A011. Cells were treated with A011in the presence or absence of3-MA for 24 h and then detected by CCK-8 assay. (E) The autophagosom-like structures in cells were detected by laser confocal microscope after A011 treatment for 12 h. Data represent mean ± SD of three different experiments. *P < 0.05, * *P < 0.01 vs Control.
3.6. A011 inhibited the activation of the PI3K-Akt-mTOR pathway and activated the PERK-eIF2α-CHOP pathway
PI3K-Akt-mTOR pathway is one of the confluences of upstream pathways that regulate cell growth, proliferation, motility, survival and autophagy. Its main downstream targets include eukaryotic translation initiation factor 4E binding protein 1 (4EBP1) and p70 ribosomal protein S6 kinase (P70S6K), which play important roles in initiating transcription and translation of related genes and regulating autophagy [44], [45]. We measured PI3K-Akt-mTOR pathway related proteins by western blot and found that the phosphorylation levels of phosphatidylinositol 3-kinase (PI3K), serine/threonine kinase (AKT), mammalian target of rapamycin (mTOR), P70S6K and 4EBP1 proteins in MCF-7 cells were significantly decreased in a concentration dependent manner after 24 h of A011 intervention (Fig. 5 A). PERK-eIF2α-CHOP signaling pathway is the key ER stresspathway. Severe or sustained ER stress can cause irreversible cell damage, lead to up-regulation of CCAAT/enhancer-binding protein homologous protein (CHOP) expression and ultimately activate caspase cascade, resulting in cell apoptosis [46], [47]. After A011 treatment, the phosphorylation levels of protein kinase RNA-like endoplasmic reticulum kinase (PERK) and eukaryotic translation initiation factor (eIF) 2a proteins in MCF-7 cells were significantly increased, and both the expression of CHOP and cleaved-caspase-9 proteins were also significantly up-regulated (Fig. 5B).
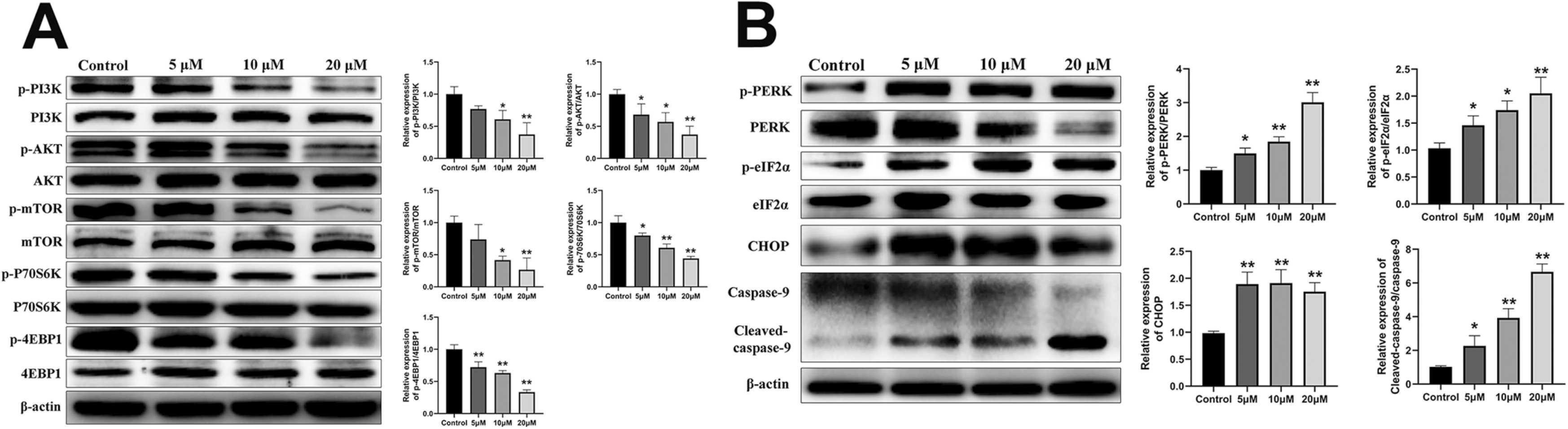
A011 inhibited the activation of the PI3K-Akt-mTOR pathway and activated the PERK-eIF2α-CHOP pathway. (A) A011 significantly decreased the expressions of phospho-PI3K, phospho-Akt, phospho-mTOR, phospho-P70S6K and phospho-4EBP1 proteins. (B) A011 significantly increased the expressions of phospho-PERK, phospho-eIF2α, CHOP and cleaved-caspase-9 proteins. Cells were treated with A011 for 24 h and then detected by western blotassay. Data represent mean ± SD of three different experiments.*P < 0.05, * *P < 0.01 vs Control.
3.7. A011 inhibits the growth of breast tumor in vivo
To evaluate the in vivo antitumor efficacy of A011, MCF-7 cells were inoculated subcutaneously to establish xenograft models. Cisplatin was used as the positive control. After 15 days of intraperitoneal injection of different doses of A011 (0, 2, 4, 8 mg/kg) or cisplatin (2 mg/kg), the tumor weight (mg) and tumor volume (mm3) growth curves of each group were measured (Fig. 6A-C). Compared to the control group, the relative growth inhibition rates of A011 at 2, 4 and 8 mg/kg were 31.8%, 42.3% and 49.2%, respectively. At a dose of 4 mg/kg, A011 had a 40% inhibitory effect on tumor growth, which is similar to the inhibitory rate by cisplatin at 2 mg/kg. Compared to the control group, the mean body weight of nude mice in the cisplatin group was significantly reduced with an average weight loss of 11.6%, while the mean body weight of nude mice in the A011 administration group was not significantly affected. In addition, the experimental animals showed no other adverse performance, indicating that the experimental animals had a good tolerance to A011 (Fig. 6D).
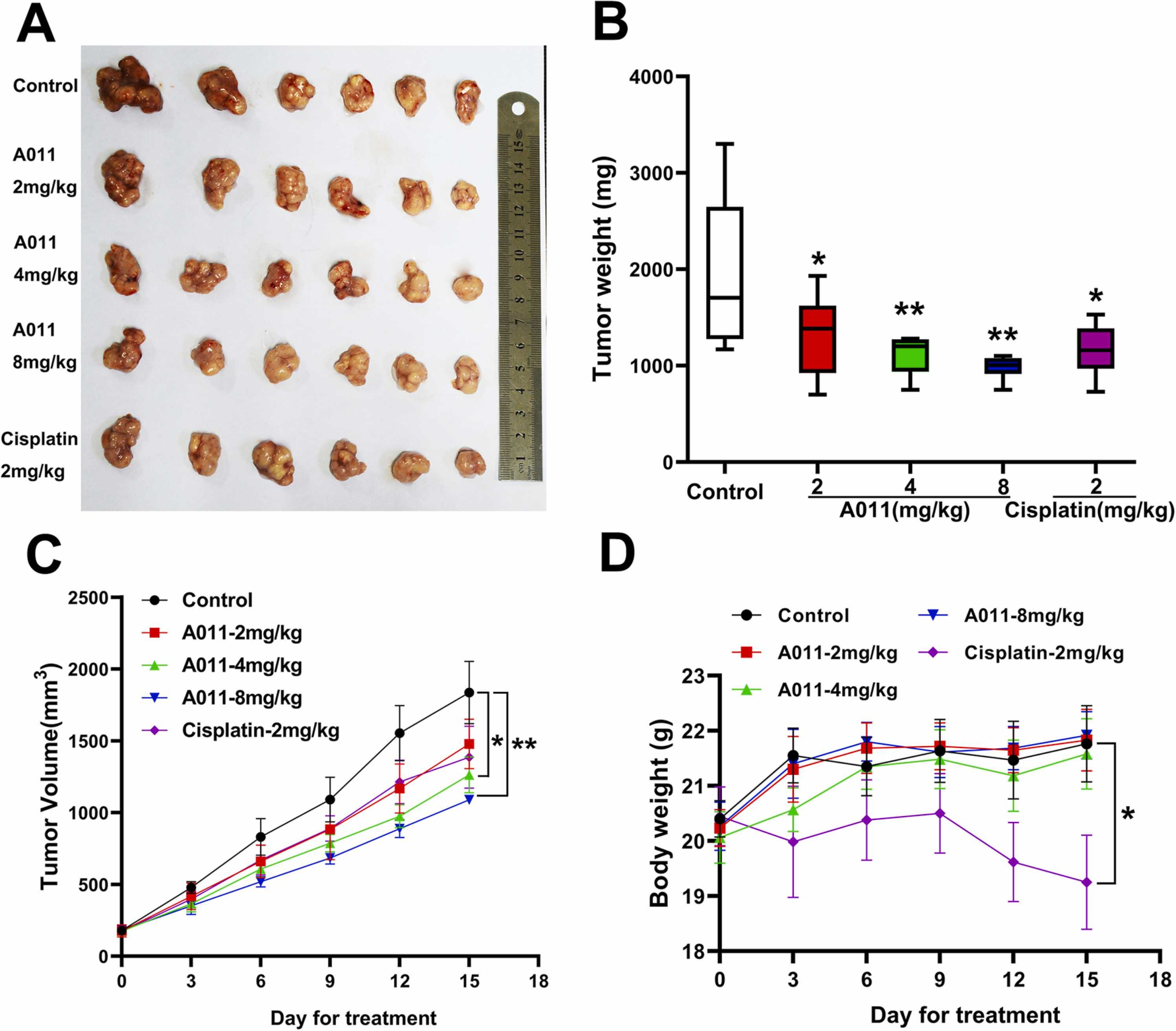
A011 inhibits tumor growth but do not reduce body weight in xenograft mouse model. Cells were transplanted in the BALB/c-nu/nu mice and treated with the tested articles indicated. (A) The representative photographs of tumors after last treatment with placebo (control), A011 at different doses, or cisplatin at 2 mg/kg; (B) Mean tumor weight after excising from the mice on the 15th day after implantation; (C) The changes in tumor volume over time after the MCF-7 cells implantation with placebo (control), A011 at different doses, or cisplatin at 2 mg/kg; (D) The changes in mean body weight of nude mice after treatment with placebo (control), A011 at different doses, or cisplatin at 2 mg/kg. Data represent mean ± SD, n = 6. *P < 0.05, * *P < 0.01 vs Control.
3.8. A011 induced autophagy and apoptosis of tumor cells but showed no obvious toxicity toward main organs in vivo
To evaluate the effects of A011 on the liver, kidney and tumor tissues of nude mice, HE staining analysis were performed on the related tissues. Histological analysis of the liver and kidney tissues showed no significant difference between the control group and the A011 treatment group, while the nude mice in the cisplatin group showed some mild swelling in liver cells and mild congestion in the renal tubuleinterstitial. As for the tumor tissue, about 40% of the tumor tissues in the control group were necrotic, while about 70% and 60% of the tumor tissues, respectively, in the A011 treatment group and the cisplatin group were necrotic. The necrotic lesions of the tumor tissues were pink, with loose structure and large nuclear to cytoplasm ratio (Fig. 7 A). TUNEL staining showed that apoptosis was significantly increased in the A011 treatment group compared to the control group (Fig. 7B and Supporting Information Fig. S1). Immunohistochemically staining of LC3 protein, a marker of autophagy in cells, showed that LC3 positive expression cells were observed in the control group. With the increase of A011 administration dose, the proportion of LC3 positive expression cells gradually increased, resulting in dark brown appearance (Fig. 7 C and Supporting Information Fig. S1). The above results suggest that A011 induces autophagy and apoptosis in tumor cells but has no obvious toxicity in main organs in vivo.

A011 induced autophagy and apoptosis of tumor cells but showed no obvious toxicity toward main organs in xenograft mouse model. Cells were transplanted in the BALB/c-nu/nu mice and treated with the tested articles indicated. (A) Hematoxylin-eosin staining analysis of liver, kidney and tumor tissue of nude mice in A011 high-dose group, cisplatin group and control group (200x or 400x); (B) TUNEL staining to evaluate the proportion of apoptotic cells in tumor tissue (100x or 200x); (C) Immunohistochemically staining to evaluate the expression of LC3 (100x or 200x). Data represent mean ± SD, n = 6. Scale bar = 100 µm or 50 µm.
4. Discussion
σ2 receptor is highly expressed in many types of rapidly proliferating cancer cellsand is considered a candidate target for cancer imaging and treatment of common proliferative tumors [48], [49]. The biggest disadvantage of traditional anti-tumor drugs is high toxicity and low specificity. Accumulating evidence has demonstrated that σ2 ligands as anti-tumor agents have the characteristics of high efficiency and low toxicity [36], [50]. Through cytotoxicity experiments on several common types of cancer cells, we found that A011, as a ligand of σ2 receptor, had the best activity against breast cancer MCF-7 cells, which was confirmed to be a cell line with low expression of σ1 receptor and high expression of σ2 receptor [51]. Subsequent in vitro experiments indicated that A011 exhibited excellent growth inhibition against breast cancer cells. Furthermore, A011 showed obvious antitumor effect and relatively low toxicity to normal tissues in vivo.
ER is the main storage site of Ca2+, and any changes in its concentration may affect ER homeostasis [52]. Previous studies have shown that a lot of σ2 ligands stimulate intracellular Ca2+ release from its storage [32]. In this study, we further explored whether A011 can cause ER stress. During ER stress, accumulating of unfolded proteins in the ER lumen causes PERK to be released from glucose regulated protein 78 (GRP78) and then to be activated, leading to the phosphorylation of eIF2a. Subsequently, phosphorylated eIF2a induces translation of activating transcription factor 4 (ATF4) that promotes the expression of the apoptotic factor of CHOP [46], [47]. CHOP is a specific transcription factor of ER stress, and its high expression is considered to be a marker of ER stress-induced apoptosis [53]. When ER stress exceeds the threshold, CHOP transcription is activated by the UPRpathway, in which the PERK-eIF2α-ATF4-CHOP pathway is the main signaling pathway that induces its transcription. As a major apoptotic factor of ER stress, CHOP can induce cell apoptosis through a variety of pathways [54]. Our study showed that A011 indeed induced a rapid increase of cytosolic free Ca2+ levels in MCF-7 cells. Subsequent pharmacological studies found that after A011 treatment for 24 h, the phosphorylation levels of PERK and eIF2a proteins were significantly increased, and the expression of CHOP and cleaved-caspase-9 protein were both significantly up-regulated. Taken together, these results suggest that A011 activates PERK-eIF2α-CHOP signaling pathway, which in turn induces the caspase cascade and ultimately leads to apoptosis.
ROS can be produced in the ER and mitochondria and plays a key role in many cellular processes. ER stress and mitochondrial oxidative stress are interconnected and usually occur simultaneously or successively. During ER stress, disulfide bondbreakage leads to ROS accumulation, oxidative stress and mitochondrial dysfunction. Similarly, oxidative stress disrupts the ER redox state, resulting in disruption of the proper disulfide bond formation and protein folding [54]. In fact, ER stress and oxidative stress can aggravate each other to interfere with cell function and activate pro-apoptotic signals. We found that after A011 intervention for 4 h, the level of intracellular ROS in MCF-7 cells was significantly increased along with the decrease of mitochondrial membrane potential (Supporting Information Fig. S2), whereas the application of vitamin E reduced the elevated ROS level in cells induced by A011. Moreover, CCK-8 assay showed that vitamin E treatment could reverse the A011-mediated proliferation toxicity in MCF-7 cells to a certain extent. This suggests that the increase of intracellular ROS level is one of the mechanisms of A011 promoting apoptosis.
Autophagy maintains intracellular homeostasis by degrading misfolded proteins, damaged organelles, and cytoplasmic components [55]. Studies have shown that ER stress stimulates the assembly of the pre-autophagosomal structure [56]. In addition, the PI3K-Akt-mTOR pathway also plays an important role in regulating the autophagic response of cells in response to change ROS levels [57]. Autophagy is initiated by the UNC-51-like kinase (ULK) complex which is inhibited by mTOR[58]. As a downstream target of PI3K protein, Akt acts by activating a mechanistic target of the rapamycin complex1 (mTORC1) and by arresting Atg expression [59]. High level of ROS inactivates Akt protein and promotes the expression of Atg and autophagy mediator protein Beclin1, leading to the induction of autophagy [60]. In fact, the Beclin1 protein stands at the crossroads of autophagy and apoptosis, and its activity depends upon ROS levels to a large extent. Under normal physiological conditions, Beclin1 binds to Bcl-2 protein and stays inactive. At high ROS level, Beclin1 disassociates from Bcl-2, leading to the initiation of autophagy. Furthermore, caspases cleave Beclin1 and its cleavage products can bind to mitochondrial membrane, leading to the release of pro-apoptotic factors and accelerating the apoptotic process [61], [62]. In this study, we found that after A011 intervention for 24 h, the phosphorylation levels of PI3K, Akt, mTOR, P70S6K and 4EBP1 proteins in MCF-7 cells were all significantly decreased in a concentration dependent manner. These results suggest that A011 induces autophagy by inhibiting the activation of the PI3K-Akt-mTOR pathway, which is one of the mechanisms of A011 promoting apoptosis.
Another reason for the research enthusiasm for σ2 ligands is that these compounds have shown potential in combined administration with other anticancer drugs and reverse tumor resistance. For example, Azzariti and coworkers found that PB-28 could reduce the expression of P-glycoprotein (P-gp) in MCF-7 and MCF-7/ADR cells in a concentration and time-dependent manner [63]. They also found that there was a strong synergism between PB-28 and doxorubicin by adopting either simultaneous or sequential schedules of the two drugs [63]. Riganti and coworkers found that three compounds F397, F400, and F421, derived from different classes of σ2 ligands, showed highly cytotoxic to multidrug resistant lung cancer cells in vitroand in vivo. Furthermore, these ligands potentiated the cytotoxicity induced by cisplatin [64]. Accumulating evidence has demonstrated that quite a few σ2 ligands can interact with P-gp efflux pump and inhibit the activity of P-gp [65]. Nearly half of human cancers are resistant to anticancer therapy due to overexpression of P-gp [66]. Therefore, σ2 ligands may act as single antitumor agents that are able to overcome P-gp-mediated resistance or can be co-administrated with a classic anticancer drug whose activity is hampered by P-gp overexpression. We also found that A011 has significant inhibitory effect on cisplatin-resistant lung cancer A549/DDP cells, liver cancer HepG2/DDP cells and adriamycin-resistant breast cancer MCF-7/ADR cells, and this effect seems to be related to the inhibition of P-gp (data not shown). In addition, A011 has a high affinity for σ2 receptor and can rapidly increase intracellular Ca2+ levels, inducing ER stress and resulting in mitochondrial dysfunction. Therefore, interference with energy supply may also be one of the mechanisms by which A011 exerts its effect against drug-resistant cancer cells. More research is needed to clarify the mechanism by which A011 reverses multidrug resistance.
5. Conclusions
In this study, we elucidated that A011, a new compound containing the affinity group of 6, 7-dimethoxy-1,2,3,4-tetrahydroisoquinoline, had a high affinity and selectivity for σ2 receptor and exhibited significant anti-breast cancer activity and low toxicity to normal tissues in vivo and in vitro. Molecular pharmacology studies revealed that A011 can significantly increase intracellular ROS and Ca2+ levels, block cell cycle, induce endoplasmic reticulum stress and autophagy, inhibit PI3K-Akt-mTOR pathway and activate PERK-eIF2α-CHOP pathway. A011 might be potentially useful as a therapeutic drug for treating breast cancer. A schematic diagram summarizing the mechanisms of anticancer by A011 was showed below (Fig. 8).
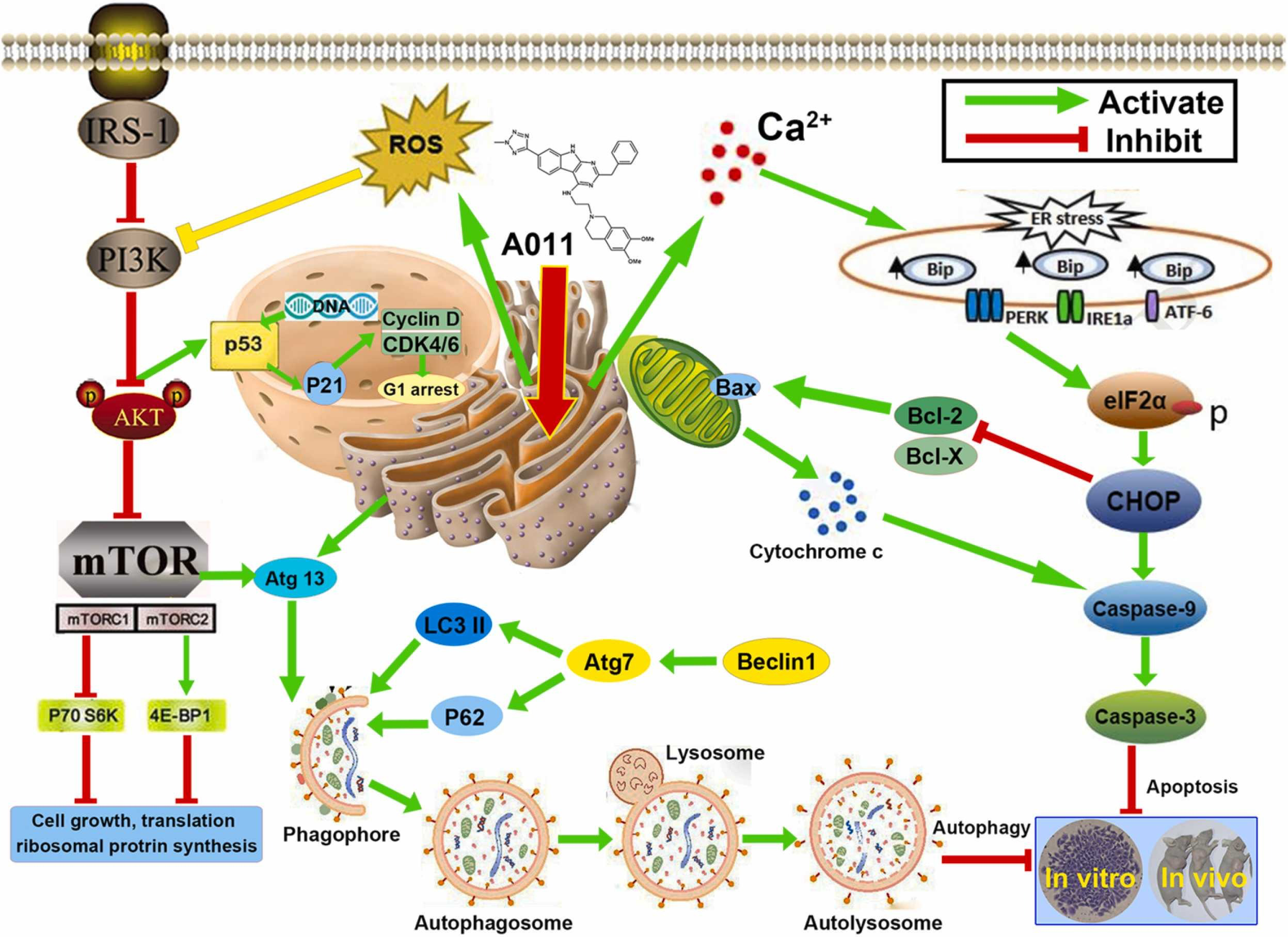
The schematic diagram summarizing the anticancer mechanism of A011. A011 increased intracellular ROS and Ca2+ levels, induced endoplasmic reticulum stress and autophagy, inhibited PI3K-Akt-mTOR pathway and activated PERK-eIF2α-CHOP pathway, thus leading to cell cycle arrest and apoptosis.