
Synaptic oligomeric tau in Alzheimer’s disease — A potential culprit in the spread of tau pathology through the brain
By Martí Colom-Cadena, Caitlin Davies, Sònia Sirisi, Ji-Eun Lee, Elizabeth M. Simzer, Makis Tzioras, Marta Querol-Vilaseca, Érika Sánchez-Aced, Ya Yin Chang, Kristjan Holt, Robert I. McGeachan, Jamie Rose, Jane Tulloch, Lewis Wilkins, Colin Smith, Teodora Andrian et al.
Excerpt from the article published in Neuron, Elsevier, May 15, 2023, DOI: https://doi.org/10.1016/j.neuron.2023.04.020
Highlights
- Oligomeric tau is present in synapses in Alzheimer’s disease cortex
- There is more oligomeric tau than phosphorylated or misfolded tau in synapses
- Synaptic oligomeric tau is present even in areas without tangles
- Oligomeric tau spreads from pre to post synapses in a mouse model
Summary
In Alzheimer’s disease, fibrillar tau pathology accumulates and spreads through the brain and synapses are lost. Evidence from mouse models indicates that tau spreads trans-synaptically from pre- to postsynapses and that oligomeric tau is synaptotoxic, but data on synaptic tau in human brain are scarce. Here we used sub-diffraction-limit microscopy to study synaptic tau accumulation in postmortem temporal and occipital cortices of human Alzheimer’s and control donors. Oligomeric tau is present in pre- and postsynaptic terminals, even in areas without abundant fibrillar tau deposition. Furthermore, there is a higher proportion of oligomeric tau compared with phosphorylated or misfolded tau found at synaptic terminals. These data suggest that accumulation of oligomeric tau in synapses is an early event in pathogenesis and that tau pathology may progress through the brain via trans-synaptic spread in human disease. Thus, specifically reducing oligomeric tau at synapses may be a promising therapeutic strategy for Alzheimer’s disease.

Introduction
Of the neuropathological hallmarks of Alzheimer’s disease (AD), synapse loss is the strongest correlate of cognitive decline,1,2,3 and progression of tau pathology through the AD brain closely correlates with synaptic loss and cognitive symptoms.4,5 Tau protein binds synaptic vesicles and is thought to have physiological roles in synapses.6 In animal and neuronal culture model systems, accumulation of hyperphosphorylated and misfolded tau within synapses causes synaptic dysfunction and synapse loss. 7,8,9,10,11,12,13
While neurofibrillary tangles (NFTs) were historically considered neurotoxic lesions, data over the past decades indicate that the tau fibrils in tangles are relatively inert and that soluble oligomeric forms of tau are more toxic to synapses and neurons in mouse models of tauopathy.14,15 Much less is known about synaptic tau and in particular synaptic oligomers in human brain, which is an important knowledge gap to address in order to develop tau-directed therapeutics to protect synapses from degeneration. Phosphorylated and misfolded tau have previously been observed in human AD synapses using biochemical and imaging methods.12,16,17 There is a small amount of evidence from human cases suggesting that diffuse forms of tau precede NFT formation,18,19 that those diffuse forms are mainly oligomeric species,20,21 and that oligomeric tau is important for seeding activity.22 Both high seeding activity and high concentrations of oligomers have been linked to worse cognitive outcomes.22,23
In addition to synaptotoxic effects, synaptic localization of tau is potentially important for propagation of tau pathology through brain networks. Seminal observations of the patterns of tau pathology postmortem by Braak and Braak24 and more recent observations using tau ligands for positron emission tomography (PET)25,26 demonstrate that NFTs accumulate in a stereotypical spatiotemporal sequence beginning in the brain stem and trans-entorhinal cortex and progressing through synaptically connected neural circuits. Because tau is secreted by presynaptic terminals in an activity-dependent manner27 and misfolded tau can act as a seed to initiate pathological changes in naive tau in vitro and in vivo,28,29 a likely method of pathological propagation is via synaptic connections. It is also possible that anatomically connected brain regions are sequentially vulnerable to tau. A recent analysis of tau PET imaging and postmortem data indicates that local replication, rather than spreading between brain regions, may be more closely related to regional tau pathology.30 These two ideas are not mutually exclusive, and indeed the local accumulation of tau may at least partially reflect trans-synaptic spread in local circuits, which one might predict would occur faster because of the shorter axons of local connections.
We and others have previously demonstrated trans-synaptic tau spread in model systems including transgenic mice expressing human tau in the entorhinal cortex and viral-mediated tau expression.31,32,33,34,35 These studies prove that trans-synaptic tau spread is biologically possible. However, one limitation is that it might have been artificially induced by overexpression in models, and this phenomenon has not been conclusively demonstrated in human brain. Here, we use sub-diffraction-limit optical imaging of synapses with multiple techniques and multiple tau antibodies (detecting oligomeric, misfolded, and phosphorylated tau) to explore in human postmortem brain whether there is evidence of tau present in both the pre- and postsynaptic sides of the same synapses, which could be due to trans-synaptic spread. Furthermore, we characterize tau pathology in detail to determine whether synaptic tau accumulates in areas without substantial NFTs.
Results
Excitatory synapses are lost in Alzheimer’s disease
Using human postmortem brain samples prepared for array tomography (Table 1), we first replicated the observations from previous studies where synapse loss has been observed in AD.1,2,3,36,37 Using array tomography, we imaged 1,315,583 individual excitatory synaptic pairs in inferior temporal cortex (BA20/21). Synaptic pairs were defined as a synaptophysin-positive presynaptic puncta with a PSD95 puncta within 0.5 μm (distance between centroids). This distance was chosen based on electron microscopy data and analysis of synaptic pairs in this array tomography dataset with differing cut-off distances (Figures S1A and S1B). We observed a 1.25-fold decrease in excitatory synapse density in AD when compared to controls (Control median = 4.29 × 108 ± 7.49 × 107 [n = 12]; AD median = 3.45 × 108 ± 9.99 × 107 [n = 20]; β = −5,314.01; p = 0.023; Figures 1A and 1B ). Next, we investigated the relationship between pathological aggregates and synaptic loss in the immediate environment. To do this, regions of interest were chosen at 63× magnification with either a neurofibrillary tangle, neuritic plaque, or no aggregate in the center of the field of view (140.6 × 106.6 μm area; Figure S1C). A decrease in synapse density was observed around clumps of dystrophic neurites (DNs) containing tau that surround amyloid aggregates in neuritic plaques (Control β = 3,862; p = 0.005; AD β = 3,862; p = 0.00537,38). In contrast to DNs, the presence of NFTs was not associated with a decreased number of excitatory synapses in the immediate environment (Figures 1A and 1C).
| BBN | SSD | Clinical diagnosis | APOE genotype | Age (years) | Sex | PMI (h) | Brain weight (g) | Brain pH | Braak stage | Thal phase | Study |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BBN_19686 | SD063/13 | control | 3/3 | 77 | F | 75 | 1320 | 6.5 | I | I | 1,9 |
| 001.26495 | SD024/15 | control | 3/3 | 78 | M | 39 | 1290 | 6.17 | I | 0 | 1,2,5,7,8,9 |
| 001.28402 | SD051/15 | control | 3/3 | 79 | M | 49 | 1503 | 6.33 | I | 2 | 1 |
| 001.28406 | SD001/16 | control | 3/3 | 79 | M | 72 | 1437 | 6.13 | II | 2 | 1,2,5,6,7,8,9 |
| 001.28793 | SD017/16 | control | 3/3 | 79 | F | 72 | 1219 | 5.95 | II | 1 | 1,2,5,6,7,8,9 |
| 001.28797 | SD025/16 | control | 3/3 | 79 | M | 57 | 1301 | 6.11 | 0 | 0 | 1,2,5,6,7,8 |
| 001.32577 | SD002/18 | control | 3/3 | 81 | M | 74 | 1313 | 6.07 | II | 3 | 1 |
| 001.35215 | SD008/19 | control | 3/3 | 82 | M | 40 | 1246 | 6.01 | I | NA | 1 |
| 001.35549 | SD030/19 | control | 3/3 | 82 | M | 56 | 1326 | 6.01 | I | 1 | 1 |
| 001.29082 | SD031/16 | control | 3/4 | 79 | F | 80 | 1339 | 5.96 | III | 5 | 1,2,5,6,7,8 |
| 001.34131 | SD029/18 | control | 3/4 | 82 | M | 95 | 1472 | 5.97 | IV | 3 | 1,2,5,6,7,8,9 |
| 001.35181 | SD003/19 | control | 3/4 | 82 | M | 49 | 1,496 | 6.12 | II | 2 | 1,2,5,6,7,8,9 |
| 001.28794 | SD018/16 | control | 2/3 | 79 | F | 61 | 1,289 | 5.76 | I | 0 | 2,5,7,8,9 |
| 001.29086 | SD034/16 | control | 3/3 | 79 | F | 68 | 1,468 | 6.2 | 0 | 1 | 2,5,6,7,8,9 |
| 001.35866 | SD046/19 | control | NA | 83 | F | 78 | 1,158 | 5.96 | IV | 4 | 2,5,6,7,8,9 |
| 001.36435 | SD013/21 | control | 3/3 | 84 | M | 95 | 1,222 | 6.00 | II | 1 | 3 |
| 001.35511 | SD025/19 | control | 2/3 | 86 | M | 123 | 1,553 | 6.17 | II | 1 | 3 |
| 001.36135 | SD012/20 | control | 3/3 | 84 | F | 30 | 1,214 | 5.93 | II | 1 | 3 |
| 001.36809 | SD023/21 | control | 3/3 | 85 | M | 3 | 1,400 | 6.10 | II | 1 | 3 |
| BBN_24527 | SD056/14 | AD | 3/3 | 81 | M | 74 | 1,160 | 6.1 | V | 4 | 1,2,5,6,7,8,9 |
| 001.28410 | SD005/16 | AD | 3/3 | 62 | F | 109 | 1,029 | 6.04 | VI | 5 | 1 |
| 001.28771 | SD010/16 | AD | 3/3 | 85 | M | 91 | 1,183 | 5.95 | VI | 5 | 1,2,5,6,7,8,9 |
| 001.29081 | SD030/16 | AD | 2/3 | 90 | F | 110 | 943 | 6.06 | VI | 5 | 1 |
| 001.32929 | SD012/18 | AD | 3/3 | 85 | F | 80 | 1,354 | 6.03 | VI | 5 | 1,2,5,6,7,8,9 |
| 001.35182 | SD004/19 | AD | 3/3 | 66 | M | 49 | 1,225 | 6.12 | VI | 5 | 1,9 |
| BBN_24322 | SD049/14 | AD | 3/4 | 80 | M | 101 | 1,410 | 6.10 | VI | 5 | 1,2,5,7,8 |
| BBN_24526 | SD055/14 | AD | 3/4 | 79 | M | 65 | 1,300 | 6.05 | VI | NA | 1 |
| BBN_24668 | SD058/14 | AD | 3/4 | 96 | F | 61 | 1,082 | 6.11 | VI | NA | 1 |
| BBN_25739 | SD014/15 | AD | 3/4 | 85 | F | 45 | 1,375 | 5.77 | VI | 5 | 1,2,5,6,7,8,9 |
| 001.26718 | SD040/15 | AD | 3/4 | 78 | M | 74 | 1,367 | 6.13 | VI | 5 | 1,2,5,6,7,8,9 |
| 001.29135 | SD027/16 | AD | 3/4 | 90 | M | 73 | 1,275 | 6.44 | VI | 3 | 1 |
| 001.29521 | SD035/16 | AD | 3/4 | 95 | M | 96 | 1,221 | 6.08 | VI | 5 | 1 |
| 001.29695 | SD004/17 | AD | 3/4 | 86 | M | 72 | 1,300 | 6.1 | VI | 5 | 1,2,5,7,8,9 |
| 001.30142 | SD020/17 | AD | 3/4 | 88 | F | 112 | 1,054 | 5.96 | VI | 5 | 1,2,5,6,7,8,9 |
| 001.30883 | SD034/17 | AD | 3/4 | 61 | F | 69 | 1,060 | 6.15 | VI | 5 | 1 |
| 001.30973 | SD039/17 | AD | 3/4 | 89 | F | 96 | 1,210 | 6.03 | VI | 5 | 1,2,5,6,7,8,9 |
| 001.33636 | SD017/18 | AD | 3/4 | 93 | M | 43 | 1,117 | 5.92 | VI | 5 | 1 |
| 001.35096 | SD037/18 | AD | 3/4 | 72 | M | 103 | 1,209 | 6.18 | VI | 5 | 1,2,5,6,7,8 |
| 001.35535 | SD028/19 | AD | 3/4 | 83 | F | 95 | 1,400 | 6.55 | VI | 4 | 1 |
| 001.35564 | SD031/19 | AD | 3/4 | 90 | F | 52 | 1,075 | 5.99 | VI | 5 | 3 |
| 001.36297 | SD022/20 | AD | 3/4 | 84 | M | 87 | 1,163 | 5.82 | VI | 5 | 3 |
| 001.36346 | SD027/20 | AD | NA | 90 | F | 60 | 932 | 6.1 | VI | 5 | 3 |
| AD | NA | 90 | F | 18 | 1,110 | NA | V | 5 | 4 |
Demographic, neuropathological, and genetic characteristics of human subjects
AD, Alzheimer’s disease; APOE, Apolipoprotein E; BBN, Medical Research Council Brain Bank number; SD, Sudden Death Brain Bank number; NA, not available; PMI, postmortem interval. Study number as included in Table S1.
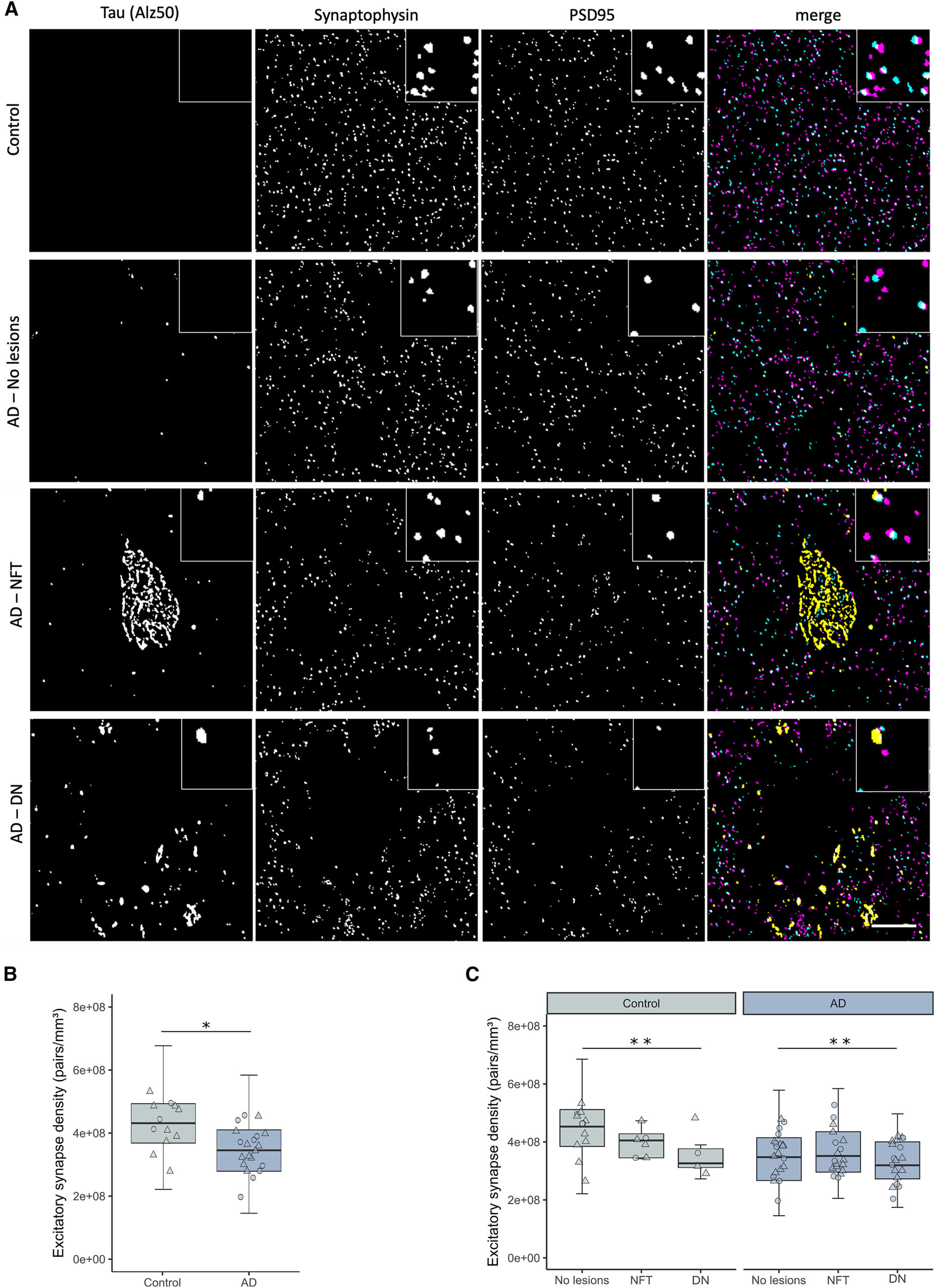
Excitatory synapse loss in BA20/21 of AD cases and its relationship to hallmark tau aggregates
A) Representative single 70-nm-thick segmented images of a control case and AD cases with no hallmark tau aggregates (AD-no lesions), a neurofibrillary tangle (AD-NFT), or dystrophic neurite clumps around neuritic plaques (AD-DN). In merged composite images, synaptophysin presynaptic terminals are shown in magenta, PSD95 postsynaptic terminals in cyan, and misfolded tau (Alz50) in yellow.
(B) Excitatory synapse density was decreased in AD cases when compared to controls.
(C) The quantification of synaptic terminals split by the presence of tau aggregates. Boxplots show quartiles and medians calculated from each image stack. Data points refer to case means (females, circles; males, triangles). Analysis was with linear mixed effects models, including diagnostic group, sex, and pathology in the image (no effect of sex). Scale bar represents 10 μm. Insets, 5 × 5 μm. NFT, neurofibrillary tangle; DN, dystrophic neurite. ∗p < 0.05, ∗∗p < 0.01. See also Figure S1 and Table S1.
Oligomeric, misfolded, and phosphorylated tau species accumulate in postsynaptic terminals of AD cases
We next investigated the presence of tau at postsynaptic terminals. Three species of tau were studied: oligomeric (T22 antibody, characterized by Lasagna-Reeves 21), misfolded (Alz50 antibody), and Ser202, Thr205 phosphorylated (AT8 antibody) (Figures 2A–2E and S2A–S2I). All three tau species were found in postsynaptic terminals of AD cases, with a higher presence of oligomeric tau stained with T22 than the other markers (Figure 2F). Control cases also had a small amount of postsynaptic tau pathology. Interestingly, T22 and AT8 were often found in the same postsynapses, while Alz50 was largely found in different synapses of AD cases (Figure 2G). There was a stronger correlation between postsynaptic tau species in AD than control cases (Figure 2G).
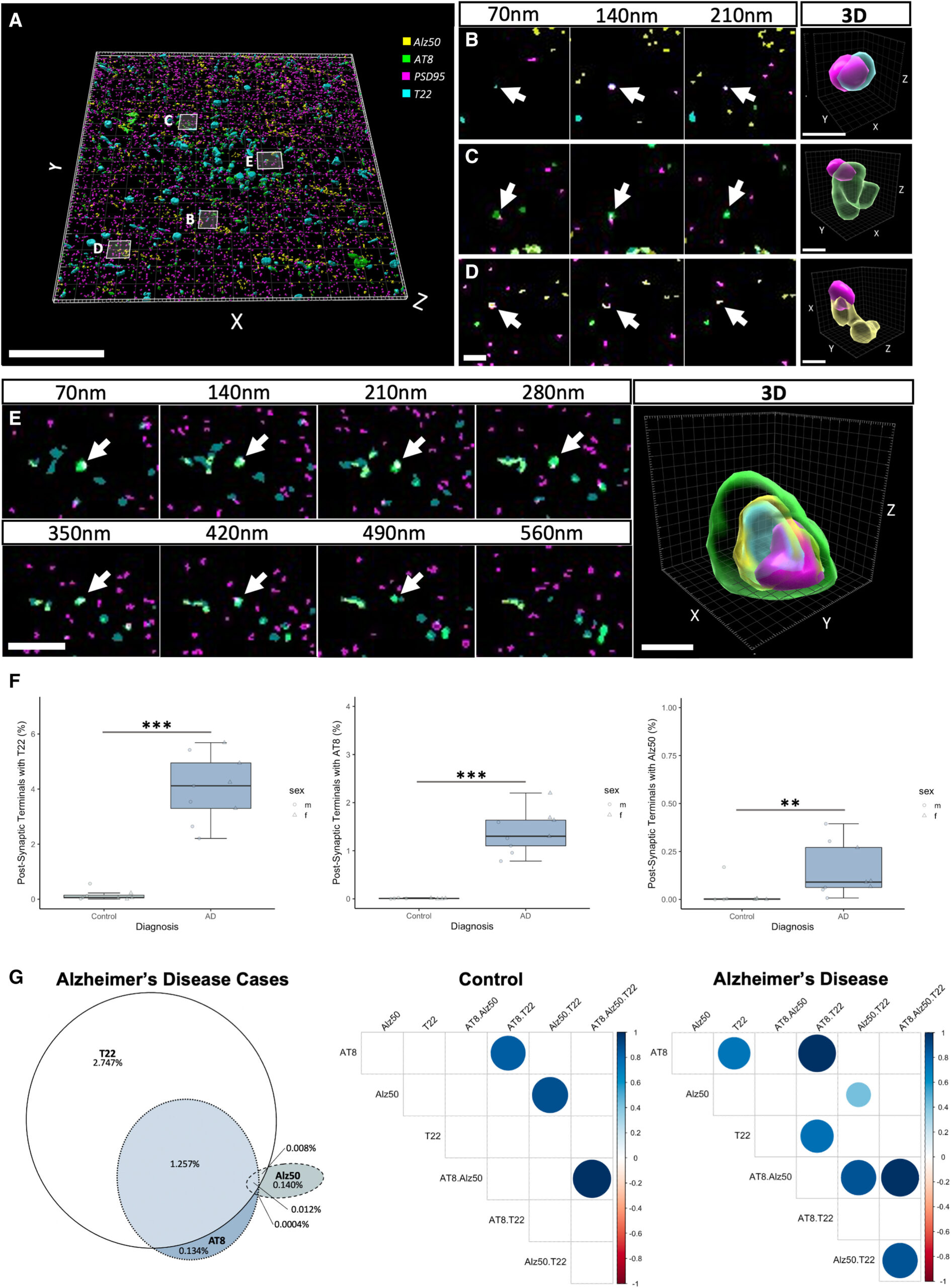
Different tau species accumulate in postsynaptic terminals
(A) An aligned and segmented three-dimensional image stack from an AD case stained for oligomeric tau (T22, cyan), misfolded tau (Alz50, yellow), Ser202, Thr205 phosphorylated tau (AT8, green), and postsynaptic densities (PSD95, magenta).
(B–E) All three types of tau can be observed in postsynapses, as shown in serial sections and three-dimensional reconstructions of individual synapses (B–E).
(F) Quantification (F) reveals increases in synaptic terminals containing tau in AD compared to control. Boxplots show quartiles and medians calculated from each image stack. Data points refer to case means (females, circles; males, triangles). ∗∗∗p < 0.001, ∗∗p < 0.01.
(G) A Venn diagram of the percentage of synapses that contain tau staining in AD cases (G, left) reveals that T22 is found in more synapses than the other species and that it colocalizes often with AT8. Alz50 colocalizes less with the other species. Correlation plots (G, right) show differences (p < 0.01) between markers in control and AD cases. For example, synaptic T22 correlates with synaptic AT8 in AD but not in control cases. Scale bars represent 20 μm in (A), 2 μm in left panels of (B)–(E), and 0.5 μm in 3D reconstructions in (B)–(E). See also Figure S2.
PSD95 puncta size did not differ between control and AD cases (Figure S2J). However, there were differences in PSD95 volume when tau species were present (ANOVA after linear mixed effects model F[3,102.3] = 6.85, p < 0.001). All tau species had a tendency to colocalize with bigger postsynaptic terminals, with T22 being the highest observed increase, especially in control cases. We observed similar results when looking at PSD95 signal intensity as a proxy for protein levels. When looking at all postsynaptic terminals, signal intensity did not differ between control and AD cases (Figure S2K). Again, there were differences when looking at the presence of different tau species and signal intensity (ANOVA after linear mixed effects model F[3,98.3] = 8.97, p < 0.001). There was an overall increase of postsynaptic intensity when each tau species was present, with T22 being the highest observed increase in control cases. Taken together, both the PSD95 size and signal intensity seem to be related to the presence of tau species, with oligomeric tau (T22) associated with the highest increases.
The presence of tau at the synapses was confirmed with other high-resolution techniques. Immunogold electron microscopy was performed in 4 control and 3 AD cases. In all cases, we observe T22-positive globular-appearing oligomers associated with fibrils in neurofibrillary tangles and on fibrils in neurites. T22 immunogold staining is also observed on presynaptic vesicles and in postsynaptic terminals near the postsynaptic density (Figures 3A and S3A). Synaptic accumulation of tau was also confirmed with direct stochastic optical reconstruction microscopy (dSTORM, 1 control case and 1 AD case) (Figures 3B and S3D) and DNA points accumulation for imaging in nanoscale topography (DNA-PAINT) in an AD case (Figures 3B and S3C). Together these super-resolution imaging techniques conclusively show pathological tau in pre- and postsynaptic terminals. Finally, the presence of oligomeric tau was biochemically confirmed with western blot of synaptoneurosome preparations from AD (n = 8) and control (n = 8) cases (Figures 3C and S3B).
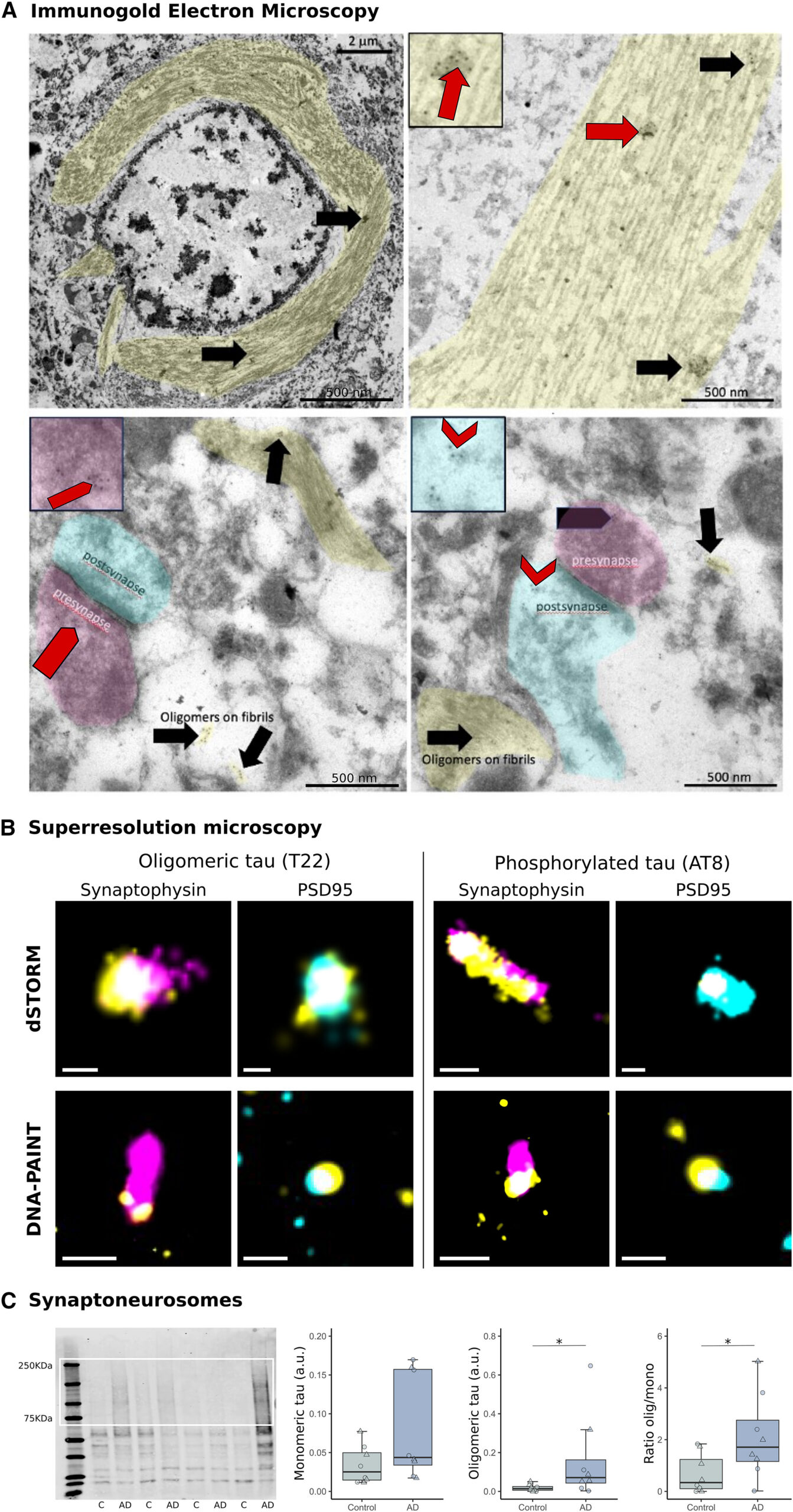
Confirmation of AD synaptic tau aggregates
(A) Immunoelectron microscopy with gold-conjugated secondary antibodies (black dots) shows T22 labels globular-appearing oligomers associated with fibrils (arrows) in neurofibrillary tangles and on fibrils in neurites (shaded yellow). T22 immunogold staining is also observed on presynaptic vesicles (pentagons, presynapses shaded magenta) and in postsynaptic terminals near the postsynaptic density (chevrons, postsynapses shaded cyan).
(B) dSTORM and DNA-PAINT were used on array tomography sections to confirm tau (yellow) and synaptophysin (magenta) or PSD95 (cyan) colocalization.
(C) Full western blot from synaptoneurosomes isolated from control or AD cases is shown at left and the quantification of oligomeric and/or monomeric bands at right. Boxplots show quartiles and medians (females, triangles; males, circles). ∗p < 0.05. C, control; SAD, Alzheimer’s disease. Scale bars EM: 500 nm (insets 250 × 250 nm), dSTORM: 100 nm, DNA-PAINT: 200 nm. See also Figure S3.
Synaptic oligomeric tau precedes the presence of neurofibrillary tangles
To determine whether oligomeric tau accumulation in synapses is an early pathological event, we next investigated the regional relationship between the presence of NFTs and the synaptic accumulation of oligomeric tau.
Temporal cortex (BA20/21) and primary visual cortex (BA17) samples from 10 AD cases and 10 controls were investigated for the presence of oligomeric tau at synaptic terminals by array tomography microscopy and for the presence of NFTs in larger fields of view by immunohistochemistry (IHC) on paraffin sections from the same cases and brain regions (Figure 4A). We found that in areas with no or low NFT counts, there was already oligomeric tau accumulating at presynaptic terminals both in control and in AD cases (Figures 4A and 4B). There was a positive correlation between NFTs and the total amount of oligomeric tau, especially in brain areas highly affected by tau pathology (AD cases at BA20/21; R = 0.81, p = 0.022; Figure 4C). However, the proportion of oligomeric tau at synaptic terminals was lower when more total oligomeric tau was present, especially in BA17, where there is less overt tau pathology (AD cases at BA17; R = −0.87, p = 0.0027; Figure 4D).

Synaptic oligomeric tau precedes the presence of neurofibrillary tangles
(A) Representative image of a case with spare (left panel) and high (right panel) NFT density. Top left images show tau AT8 immunoreactivity by immunohistochemistry and the inset highlights the presence of NFTs. At top right is a maximum-intensity projection of 12 consecutive 70-nm-thick sections by array tomography microscopy. Presynaptic synaptophysin-positive terminals (magenta) and oligomeric tau (T22, yellow) are shown. The inset highlights the presence of presynaptic terminals with oligomeric tau that are pointed by arrowheads in the horizontal panel of four consecutive 70-nm-thick sections.
(B) Table displaying the density of synaptic terminals that contained oligomeric tau and the NFT density per case, area, and Braak NFT stage. Highlighted in purple are the cases shown in (A).
(C and D) (C) Correlation plots between the density of NFTs and the percent volume of the image occupied by T22 and (D) correlation plots between the percent of T22 that colocalizes with presynaptic terminals and the total amount of T22. Spearman correlation results are shown in each condition. Data points refer to case means (females, circles; males, triangles). Scale bars: left, 100 μm; right, 10μm. Inset sizes: left, 160 × 160 μm; right, 10 × 10 μm. See also Figure S4.
To confirm whether synaptic oligomeric tau is an early phenotype, we compared staining of AT180, which labels tau phosphorylated at residue 231 (reported to be an early marker of tau pathology 39), with oligomeric tau in synapses. While both types of tau are more prevalent in AD than control synapses, we observe 3.3-fold more T22- than AT180-labeled presynaptic terminals in BA20/21 of AD cases (Presynaptic terminals with T22: control median = 0.297 ± 2.038% [n = 9]; AD median = 5.061 ± 3.389% [n = 9]; β = 4.15; p = 0.004; Figure S4B. Presynaptic terminals with AT180: control median = 0.0219 ± 0.697% [n = 9]; AD median = 1.534 ± 1.441% [n = 9]; β = 1.537; p = 0.01; Figure S4C).
Indirect evidence of transsynaptic transmission of tau aggregates
The finding of tau accumulating at synapses of AD cases even in areas with little tau pathology is in line with the hypothesis that tau may spread trans-synaptically. To further explore this possibility, the subset of tau-containing excitatory synaptic pairs was explored for pre- and postsynaptic locations of tau (Figure 5A). Oligomeric tau was differentially located in the synaptic compartments (ANOVA after linear mixed effects model F[2,88] = 73.61, p < 0.0001); it was more commonly found in presynaptic terminals only, with a gradient toward postsynaptic localization (Figure 5B). In inferior temporal cortex, BA20/21, we confirmed that misfolded tau antibody (Alz50) was also found in both sides of the synapse (Figures S5A–S5E). Finally, to overcome the descriptive nature of postmortem studies, we investigated transsynaptic spreading of oligomeric tau in an animal model. We previously demonstrated that human tau can spread from pre- to postsynaptic terminals in a mouse model with overexpression of human P301L mutant tau and synaptophysin tagged with GFP in entorhinal cortex.34. Here, tissue from this same mouse model was stained with T22 to show that oligomeric tau can spread from pre- to postsynaptic terminals in mammalian brain (Figure S5F).
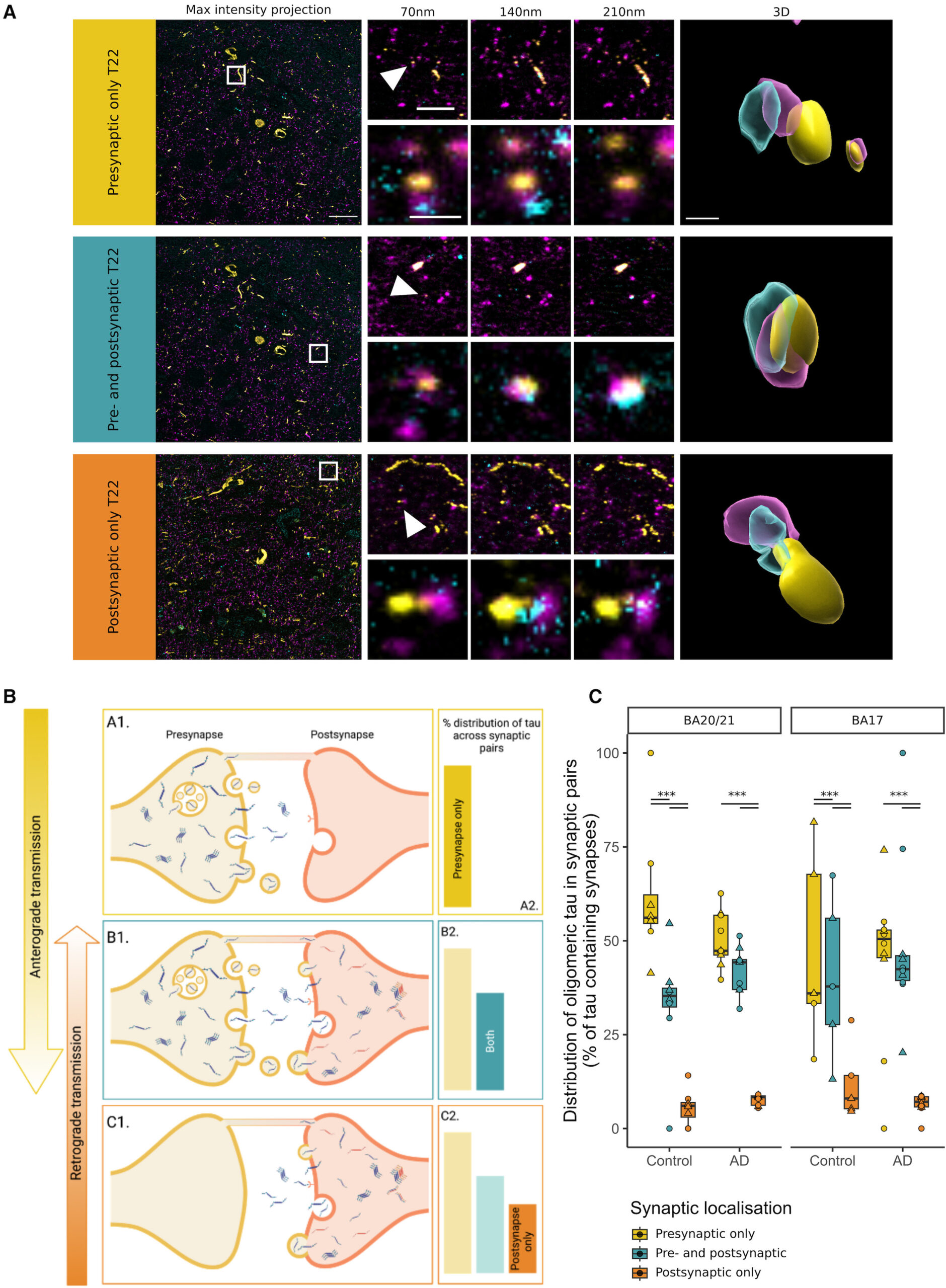
Insights into transsynaptic tau transmission
(A) Raw images from array tomography show the presence of pre- (magenta) and post- (cyan) synaptic terminals with oligomeric tau (yellow). The first column shows a maximum-intensity projection of raw images with a white box outlining the inset shown at top right of every image. Three consecutive 70-nm-thick sections are shown for the inset, and the synapse pointed by the arrowhead is shown below and at the last column in a 3D reconstruction.
(B) Diagram of the possible conditions found in tau synaptic distribution.
(C) Distribution of tau from pre- to postsynaptic terminals by diagnosis and brain region. Boxplots show quartiles and medians calculated from each image stack. Data points refer to case means (females, circles; males, triangles). Analysis was with linear mixed effects models including diagnostic group, sex, and pathology in the image. ∗∗∗p < 0.0001 in post-hoc emmeans test after linear mixed effects model. Scale bars: 20 μm (raw image), 5 μm (first inset), 1 μm (second inset), approximately 500 nm (3D). See also Figure S5.
Discussion
In the present work, we show that oligomeric, phosphorylated, and misfolded tau can be detected within synapses of human AD cases. As far as we know, this study represents the first comprehensive visualization of oligomeric tau at synaptic terminals in human tissue. The presence of oligomeric tau at synaptic terminals was observed even in cases with low NFT pathology and represented a higher proportion of the tau burden in regions with little NFT pathology, suggesting an early synaptic localization of oligomeric tau. The distribution of oligomeric tau at synapses was asymmetric, with the highest proportion in presynapses and a gradient toward the postsynapse. This is indirect evidence for transsynaptic anterograde transmission of tau.
Synaptic loss and synaptic tau
Synaptic loss and alterations in synaptic markers represent two of the earliest findings in AD-affected brain regions and are the best pathological correlates of cognitive decline.2,40,41 Here, we used array tomography microscopy,42,43 immunogold-electron microscopy, dSTORM, and DNA-PAINT, which allowed us to resolve different tau species in single synaptic terminals.
With this approach, we first replicated the synaptic loss associated with AD, as we and others have shown with several techniques.36,37,41 Previous studies using array tomography in human postmortem brain identified synapse loss in areas surrounding amyloid-β (Aβ) plaques, with synapse loss correlating with distance from the plaque.36,37 In this study, paired presynaptic and postsynaptic terminals were reduced in AD and further reduced near neuritic plaques identified by surrounding dystrophic neurites. In contrast, we did not observe synapse loss in the direct vicinity of neurofibrillary tangles. While amyloid plaques are extracellular lesions surrounded by oligomeric Aβ that is known to be toxic to synapses, neurofibrillary tangles are intracellular lesions that are thought to be harmful to the neuron containing them or to reflect a sequestering of toxic tau. This toxicity could induce loss of synapses formed by the tangle-bearing neuron, which are typically distant from the cell body, even in the opposite hemisphere. Some of the postsynaptic dendritic spines of tangle-bearing neurons are likely within the same image stack as the soma, but many of these will also be out of frame. We thus postulate that while plaques cause local synaptic toxicity, tau-induced synaptotoxicity is more diffuse throughout the brain. Second, we found that oligomeric, phosphorylated, and misfolded tau all accumulate in synaptic terminals. We observed an increase in signal intensity and volume in tau-containing postsynaptic terminals, which may reflect compensation for the injured environment, something that has been suggested previously by others.40,44 However, we need to be cautious interpreting these data, as light microscopy techniques are sensitive, and subtle changes may affect both size and intensity. We have previously observed synaptic tau with antibodies raised to total tau, misfolded tau (Alz50), and tau phosphorylated at serine 202 and threonine 205 (AT8).11,12,13 Here, we combined them and found that oligomeric tau is the most common tau species in postsynapses. This is an important finding, as animal studies indicate oligomeric tau is particularly synaptotoxic.15 In previous postmortem studies, analysis of synaptosomes45,46,47,48,49 and synaptic fractions of homogenized tissue50 have identified tau to be localized to both synaptic compartments in control and AD tissue. However, methods involving biochemical isolation of synapses fail to consider the overall architecture of the intact tissue and may unwittingly select for certain populations of synapses. Tissue homogenization can also be damaging to synapses, allowing soluble proteins to leak in or out of the preparations. Further, this method does not generally allow for study of multiple proteins within the same synapse. Array tomography overcomes some of these limitations, offering a means for high-resolution characterization of a large number of synapses in situ in human postmortem tissue, and enables the assessment of synaptic density and protein composition.42
The oligomeric tau we detected with immunoelectron microscopy is remarkably similar in morphology to oligomers biochemically isolated from human AD extracellular vesicles observed by atomic force microscopy.51 Here we demonstrate that globular structures containing oligomeric tau are associated with fibrils and within some synapses. We also observe tau labeling of synaptic vesicles consistent with observations in Drosophila that tau binds synaptic vesicle proteins and with our previous work showing with array tomography that tau is present in presynaptic terminals.12,13 This finding was further confirmed with dSTORM and DNA-PAINT, which both demonstrated colocalization between pathological tau and presynaptic vesicle proteins with nanoscale resolution.
The finding of tau, especially oligomeric tau, accumulating at synapses may have implications for the understanding of AD pathogenesis. While many studies have focused on Aβ as the trigger for AD-related synaptic damage, tau can mediate synapse loss, both alone and in concert with Aβ.52 Most tauopathy models show synapse dysfunction and loss.7,53,54,55 And mounting evidence suggests that oligomeric, misfolded forms of tau mediate synaptotoxicity. Tau oligomers may induce morphological changes, impair plasticity, and dysregulate synaptic transmission.56,57,58,59 Oligomers also induce synapse loss and impairment in memory function, which is rescued upon reduction of oligomer levels.60,61,62 Thus, at the synapse, tau oligomers may reduce synaptic proteins, alter neuronal signaling, promote synapse loss and impair memory function.
In this study, we also found oligomeric tau in cases with no or low NFT presence. These findings are in line with evidence from human cases suggesting that diffuse and neuropil forms of tau precede NFT formation18,39 and may involve oligomeric species.20,21 This early (according to NFT burden and the well-established Braak staging) oligomeric tau was found to accumulate at synapses. The seeding activity of tau in synaptoneurosome preparations has also been found to precede the amount of hallmark pathology, as reflected by Braak stages,63 and seeding closely correlated with oligomeric tau.22 Of note, both high tau seeding and high levels of tau oligomers have been linked to worse cognitive outcomes.22,23 We also found that the proportion of oligomeric tau in presynaptic terminals was inversely correlated with the total burden of oligomeric tau. Paralleling Braak staging, these findings may imply that, i.e., while primary visual cortex does not have enough pathology to be classified as Braak stage VI, we already find oligomeric tau at synapses that may be affecting neuronal circuits.
Synaptic localization of tau may be also important for propagation of tau through the AD brain. Oligomeric tau was distributed asymmetrically across synaptic pairs. This possibly suggests a model where tau accumulates presynaptically, before release and uptake at the postsynapse, leading to tau localization in both compartments. It is unlikely that transference of pathological tau to the postsynapse from the presynaptic terminal is the only propagation mechanism. Synaptic pairs in which tau is localized to the postsynapse only could arise from retrograde transport to the postsynapse (i.e., along dendrites and spines).64 Further, tau could undergo retrograde transport across the synapse to the presynaptic site.65,66 However, considering the stepwise decrease in oligomeric tau localization observed at synaptic pairs (presynapse only > both terminals > postsynapse only), it is likely that tau predominantly traverses synapses in the anterograde direction. Oligomeric tau followed this pattern of distribution across synaptic pairs in both control and AD brain. However, the proportion of synapses with tau was significantly increased in AD brain, indicating that although the mode of spread may be the same, propagation likely occurs at greater frequency in AD. While these findings gave us insights into the pathogenesis of AD and can have implications in therapeutical approaches, we need to be cautious because of the inherent descriptive cross-sectional nature of postmortem studies.
Conclusions
In the present work we found that oligomeric, misfolded, and phosphorylated tau species are found at synaptic terminals of AD brains even when NFT burden is low. Given the potential early nature of these events in AD pathogenesis, we believe that these findings support the notion of multiple target- and synaptic-based approaches for therapeutic interventions