
Targeting Sigma Receptors for the Treatment of Neurodegenerative and Neurodevelopmental Disorders

By Dicson S. Malar, Premrutai Thitilertdecha, Kanokphorn S. Ruckvongacheep, Sirikalaya Brimson, Tewin Tencomnao, and James M. Brimson
Excerpt from the article published in CNS Drugs, May 2023. DOI: https://doi.org/10.1007/s40263-023-01007-6
Editor’s Highlights
- The sigma-1 receptor (σ1R) has no homology with any other mammalian protein.
- The sigma-2 receptor (σ2R) is a genetically unrelated protein with a similarly shaped binding pocket and acts to influence cellular activities similar to the sigma-1 receptor.
- In recent years, there has been a growing interest in both the σ2R and σ1R for treatments in neurological diseases.
- Despite their lack of homology, their similar binding profile and activity of the proteins mean both receptors need to be considered when designing novel drugs, since the σ1R agonists and σ2R antagonists are neuroprotective and σ1R antagonists and σ2R agonists are used in cancer study.
- The σ1R interacts with multiple other protein complexes to modulate multiple signaling pathways and influence cellular health and survival.
Abstract
The sigma-1 receptor is a 223 amino acid-long protein with a recently identified structure. The sigma-2 receptor is a genetically unrelated protein with a similarly shaped binding pocket and acts to influence cellular activities similar to the sigma-1 receptor. Both proteins are highly expressed in neuronal tissues. As such, they have become targets for treating neurological diseases, including Alzheimer’s disease (AD), Huntington’s disease (HD), Parkinson’s disease (PD), multiple sclerosis (MS), Rett syndrome (RS), developmental and epileptic encephalopathies (DEE), and motor neuron disease/amyotrophic lateral sclerosis (MND/ALS). In recent years, there have been many pre-clinical and clinical studies of sigma receptor (1 and 2) ligands for treating neurological disease. Drugs such as blarcamesine, dextromethorphan and pridopidine, which have sigma-1 receptor activity as part of their pharmacological profile, are effective in treating multiple aspects of several neurological diseases. Furthermore, several sigma-2 receptor ligands are under investigation, including CT1812, rivastigmine and SAS0132. This review aims to provide a current and up-to-date analysis of the current clinical and pre-clinical data of drugs with sigma receptor activities for treating neurological disease.
1 Introduction
1.1 The Sigma Receptors
The sigma-1 receptor (σ1R) is a 223 amino acid chaperone protein that resides at the endoplasmic reticulum (ER); it is also found at the mitochondria-associated ER membrane (MAM), and it can translocate to the plasma membrane (Fig. 1). The σ1R is coded by the SIGMAR1 gene (found on chromosome 9 in humans and chromosome 4 in mice), which comprises four exons that code for the 223 amino acid protein. The σ1R has no homology with any other mammalian protein; however, it does share some homology with the yeast ERG2, a fungal gene encoding a C8–C7 sterol isomerase from Magnaporthe grisea, Saccharomyces cerevisiae and Ustilago maydis [1], although the σ1R has no isomerase activity.
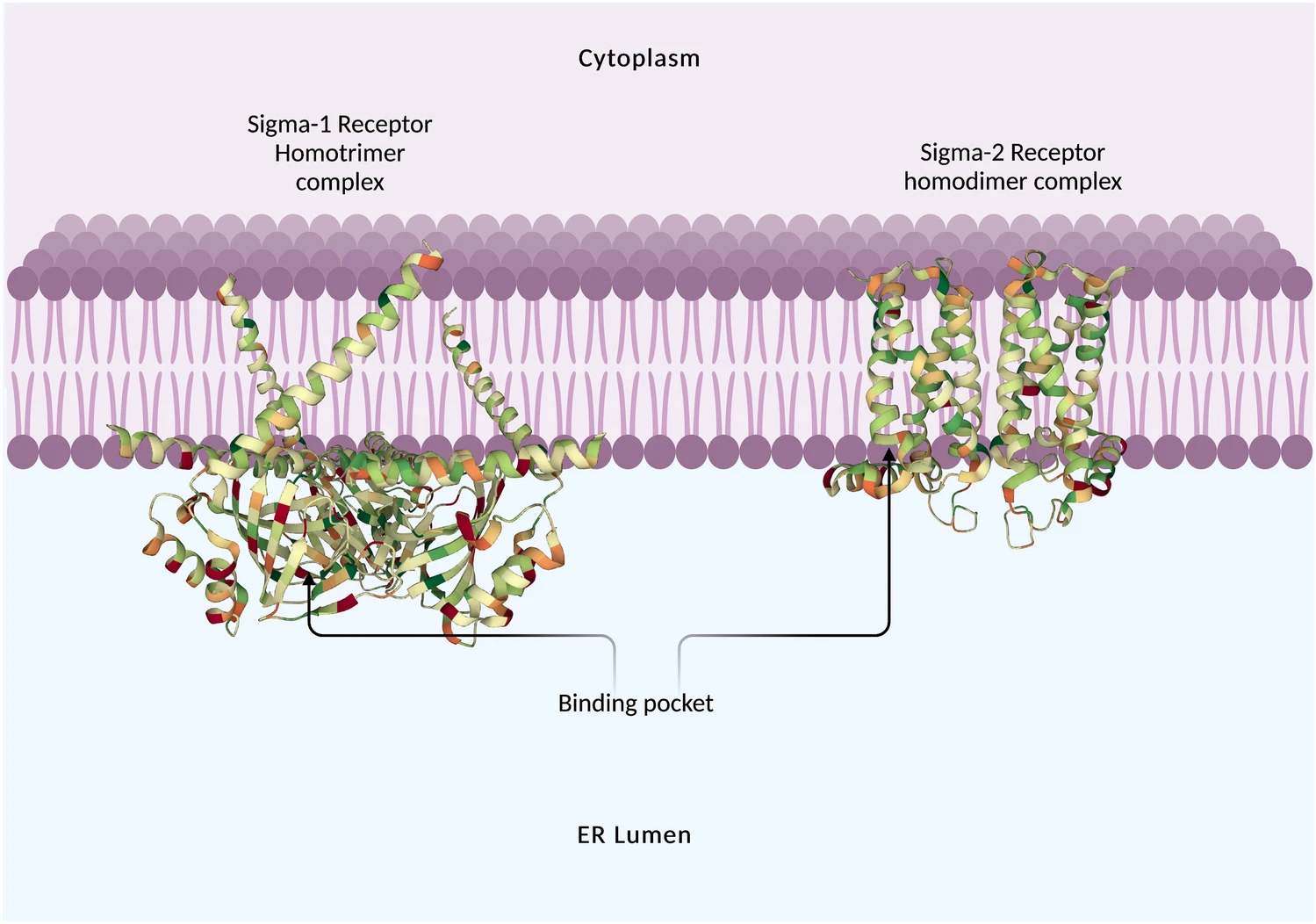
Structure of the Xenopus sigma-1 receptor homotrimer (left) bound to (+)-pentazocine (ligand removed from image) and the Bovine sigma-2 receptor homodimer (right) bound to PB28, (ligand removed from image) at the membrane of the endoplasmic reticulum.
The σ1R is concentrated in lipid-enriched microdomains at the ER and mitochondria. In NG108 neuroblastoma cells, the σ1R was shown to be important in the compartmentalization of ER-synthesized lipids. At the ER, the σ1R co-localizes with caveolin-2, a cholesterol-binding protein [2]. The σ1R also interacts with and modulates the actions of various other proteins, including the IP3R [3], ion channels [4,5,6] and the small molecule G-protein (Rac1) [7].
The σ1R has no confirmed endogenous ligand, although there are several candidates. The hallucinogen, N,N-dimethyltryptamine (DMT) [8]; neuro-active steroids such as progesterone [but also testosterone, pregnenolone sulphate and dehydroepiandrosterone sulphate (DHEA-S) bind to the σ1R] [9]; and N-alkyl amines, such as the endogenous sphingolipids [10], are all candidates as endogenous σ1R ligands.
The σ1R ligands can be divided up into several categories: the traditional agonists and antagonists that, when used at physiologically relevant doses, provide a physiological response, and the σ1R modulatory ligands, which do not provide a physiological response alone, rather they exhibit positive or negative modulatory effects when certain physiological processes are activated (such as during cellular stress or neuron firing).
The σ1R (allosteric) modulatory ligands may also act synergistically with subtherapeutic concentrations of σ1R agonists or antagonists [11, 12]. Defining σ1R ligands into these groups can be difficult since there was confusion about the identity of the sigma binding site due to a lack of specific ligands [13]. Furthermore, many early studies saw behavioural effects using what are now called σ1R agonists. In contrast, molecular pharmacological studies have shown that what are now termed σ1R antagonists produce cellular responses in calcium flux [6, 14, 15]. In general terms, σ1R agonists are ligands that have neuroprotective, mood modifying, analgesic or cognitive effects, and the antagonists mostly (there are several exceptions) have cytotoxic effects.
More specifically, laboratory tests have been suggested to define σ1R ligands into agonist or antagonist categories. One proposed, generally accepted test is to check for the dissociation of binding immunoglobulin protein (BiP), with agonists causing the dissociation whereas antagonists do not [16]. The ability to potentiate neurite outgrowth in PC12 cells indicates agonistic activity at the σ1R. Another proposed way to define σ1R ligands is the Hill slope of their binding isotherms, with a standard slope factor (slope ≈ 1) indicating an agonist and a shallow slope factor (slope ≈ 0.5) indicating an antagonist [5, 6].
The σ1R is found throughout much of the tissues in the body. However, it appears more concentrated in the central nervous system (CNS), heart, liver and eyes [17, 18]. Many cancers also overexpress the σ1R, suggesting that the σ1R plays an important role in cell survival under stressful conditions [19].
Another receptor with a similar ligand-binding profile is the sigma-2 receptor (σ2R), although it is structurally and genetically unrelated to the σ1R [20]. The σ2R has a molecular weight of 18–21.5 kDa, is located on chromosome 17 (location: 17q11.2) and was previously thought to be part of the progesterone receptor membrane component 1 (PGRMC1) [21]. A more recent study has identified the σ2R to be the transmembrane protein 97 (TMEM97), also known as meningioma-associated protein 30 (MAC30) [22]. The σ2R is an endoplasmic reticulum (ER) resident membrane protein [23, 24] and is also located in the lipid raft containing cholesterol and sphingolipids [25, 26] of the plasma membrane [27]. The structure of the σ2R has been identified using x-ray crystallography showing four transmembrane helices (Fig. 1) [28]. Although genetically unrelated to the σ1R, the binding pockets of the two proteins are remarkably similar. There are functionally comparable amino acids in similar spatial positions, which likely explains the convergent evolution of these two proteins and reveals why these two proteins bind such a similar portfolio of compounds [28].σ2Rs are found primarily in the CNS [29], breast, colon, stomach, oesophagus and lung. The σ2R is highly expressed in a large number of cancers [30], such as colorectal cancer, oral/oropharyngeal squamous cell carcinoma, breast cancer cell lines (MCF7, MDA-MB-231), neuroblastoma SK-N-SH cells [31], ovarian cancer cells [32] and prostate cancer [33].
This review aims to cover the current understanding of the sigma receptors’ role in neurodegenerative and neurodevelopmental disorder development, discuss the relevant approved or investigational drugs and critically review the available clinical data.
Literature searches were carried out (with the last search carried out in April 2023) using PubMed, Scopus and Google Scholar using search terms related to Alzheimer’s disease, Parkinson’s disease, motor neuron disease, multiple sclerosis, Rett syndrome, and developmental and epileptic encephalopathies in combination with the terms related to the σ-1/2 receptors. Clinical trials were identified via www.clinicaltrials.gov and by refining searches on PubMed only to include clinical trials. No specific time period was chosen for article publication, although more emphasis was given to recent and up-to-date articles.
1.2 Overview of Neurological Disease and the Role of the Sigma Receptors
1.2.1 Alzheimer’s Disease
Alzheimer’s disease (AD) is a neurogenerative disorder with a progressive loss of cognitive functions, commonly associated with age and neuropathology. The noticeable symptoms, such as memory loss and language problems, begin when damage occurs to neurons responsible for these cognitive functions. Other symptoms involving basic bodily functions, such as walking and swallowing, subsequently occur along with the disease progression [34]. The key components of the disease have been reported to be the accumulation of amyloid precursor protein (APP) fragment amyloid-beta (Aβ) from its overproduction or reduced clearance [35], and the downstream process in hyperphosphorylation of the protein tau, leading to cell death, neurotransmitter deficits [36], reduced synaptic strength and neurodegeneration [37], as well as induction of inflammation related to dysregulated glucose metabolism [38, 39].
The σ1R is expressed highly throughout many organs in the body; however, it is particularly concentrated in the CNS. Much research into the σ1R has focused on pain and neurotransmission [40,41,42,43,44]. In studies of normal healthy ageing, in rats or monkeys, the levels of the σ1R in the brain are maintained and possibly increased [45,46,47]. Receptor increases with healthy ageing have only been reported for σ1R and muscarinic M2 receptors. Furthermore, this increase in σ1R has been seen in human ageing studies.
Using 11C-labelled donepezil, it was shown that cardiac levels of the σ1R were elevated in aged healthy individuals [48]. This apparent increase of σ1Rs in the body appears to coincide with the age-related loss of other receptors such as the M1 and M4 muscarinic acetylcholine receptors [49], serotonin 5HT2A receptors [50] and D1/D2 dopamine (DA) receptors [51]. It has been hypothesized that the increase in σ1R expression is a compensatory mechanism to offset the loss of the other receptors [52, 53]. In the early stages of AD, there is a significant loss of σ1R expression in the brain [54, 55].
Genetic studies have identified that gene mutations in apolipoprotein E (ApoE) are related to AD risk. The ApoE2 allele is associated with a reduced risk of AD, and the ApoE3 allele is the most frequently found and is considered a neutral allele regarding AD risk. Carriers of the ApoE4 allele have an earlier onset of AD, and the amyloid plaques are more diffuse.
The σ1R gene is found on chromosome 9p13 and is approximately 7 kbp long, consisting of four exons coding for a 223 bp amino acid. There have been two reported variants of the σ1R receptor that may affect AD development. The G–241T/C–240T (rs. 1799729) in the proximal promoter region and A61C (rs. 1800866 resulted in an amino acid substitution Q2P) in the first exon [56]. The haplotype TT241-240P2 (TT-P) has been shown to express σ1R receptor protein at a level 43% lower than the allele GC-241-240Q2 [56].
Several studies have investigated the link between polymorphisms in the σ1R and the risk of AD [56,57,58,59]. A Japanese study has suggested that the TT-P haplotype is protective against AD [56]. However, this would appear to go against what is already known about the σ1R and has been contradicted by several other studies. Studies from Hungarian, Polish, Australian and Chinese populations have shown a link between the σ1R TT-P haplotype and increased susceptibility to AD [57,58,59].
Furthermore, there is evidence that there may be an interaction between the effects of the ApoE4 allele and the σ1R allele. In the Hungarian study [59], those who inherited both the TT-P haplotype and the E4 allele had a considerably higher risk for AD than those who carried only one. In the Australian/Chinese cohorts [58], the study found the Q2P polymorphism to be associated with a less severe form of AD in non-E4 allele carriers (i.e. ApoE2 or ApoE3); however, expression of the Q2P polymorphism in combination with ApoE4 had the potential to create a more severe cognitive dysfunction.
Reduction or loss of expression of the σ1R may make neuronal cells vulnerable to stress due to Aβ-mediated toxicity and damage to intracellular lipid metabolism, cell skeleton network and immune responses. These lead to neuronal cell death and contribute to the pathology of AD [60]. σ1R knockdown has been shown multiple times in primary hippocampal neurons to reduce dendritic spine formation [61,62,63].
The σ1R also plays a role in controlling tau phosphorylation. The σ1R is essential in proper tau phosphorylation and axon extension by promoting p35 turnover through the interaction of the receptor with myristic acid. Furthermore, σ1R knockout (KO) in neurons results in the accumulation of p35 due to its slower degradation. Conversely, overexpression of the σ1R results in a decrease of p35. In the σ1R-KO, neurons show shorter axons with lower densities. Interestingly the σ1R agonist myristic acid mitigates p35 accumulation, diminishes tau phosphorylation and restores axon elongation [64].
Autophagy is a cellular function that involves destroying and recycling cellular organelles and proteins, either when damaged or when the cell needs building blocks to survive. The σ1R regulates autophagy, which may be an important target for treating neurodegenerative disease (reviewed in [53]). σ1R agonists can activate autophagy, reversing some of the effects of APP overexpression [65]. The dysregulation of the autophagic process is thought to be a possible part of AD pathology [66,67,68]. This, and the fact that the σ1R is lost in the early stages of AD, could be a potential cause of, at least part of, the pathology of AD.
Calcium homeostasis is significant in AD disease development (reviewed in [69]). Vaccines/antibodies that have been trialled, which remove Aβ plaque build-up, have not stopped dementia progression despite their efficacy at removing Aβ [70, 71], suggesting that non-plaque forming Aβ-oligomers are responsible for neuron cell death [72]. Oligomers of soluble Aβ can form ion channels and dysregulate calcium influx into neurons [73]. Fibrillar Aβ aggregates may cause cell death via the induction of oxidative stress, depolarization of synaptic membranes, excessive calcium influx and mitochondrial stress [74].
The σ1R appears to play an essential role in regulating calcium homeostasis. Disrupting the σ1R in cancer cells with an antagonist such as IPAG (1-(4-iodophenyl)-3-(2-adamantyl)guanidine) or rimcazole results in a large influx of [Ca2+] into the cytosol, that can be reversed or prevented by σ1R knockdown or a σ1R agonists such as pentazocine, SKF10047 or PRE084 [6, 14, 75]. During stress, the σ1R prevents cytosolic [Ca2+] dysregulation in neuron cells [76]. The σ1R potentially helps buffer cytosolic [Ca2+] by shuttling calcium from the ER via the MAM to the mitochondria, stimulating ATP production and improving cellular health [16, 77, 78].
1.2.2 Developmental and Epileptic Encephalopathies
Developmental and epileptic encephalopathy (DEE) has recently been classified as gene-associated developmental encephalopathy without epilepsy which continues progressing even when seizures are controlled. It is then difficult to judge whether developmental or epileptic components contribute more to clinical presentation in DEE. On the other hand, developmental encephalopathy (DE) generally has only developmental impairment (e.g. intellectual disability and autistic spectrum disorder), without epileptic activity associated with regression or further slowing of development.
In contrast, epileptic encephalopathy (EE) is the epileptic activity itself contributing to severe cognitive and behavioural impairments with no pre-existing developmental delay [79]. Since most patients with DEE have aetiologies due to genetic variants, and patients with EE also have a molecular genetic basis, genetic testing to distinguish DEE becomes essential. The most frequent and studied DEE-associated genes include CDKL5, SCN1A, KCNQ2/KCNQ3, PCDH19, SCN2A/SCN8A and TSC1/TSC2. Other promising gene variants associated with an increased risk of expressing DEE comprise KCNT1, STXBP1, CACNA1A, SYNGAP1, GABRB3 and GRIN2A/GRIN2D [80]. Besides genetic variants, the causation of DEE also includes structural disorder [e.g. tuberous sclerosis complex (TSC), hypothalamic hamartomas and hemimegalencephaly], metabolic disorder (e.g. pyridoxine or biotinidase deficiency and GLUT1 deficiency) and immune disorder (e.g. Rasmussen syndrome) [81,82,83].
The current DEE treatments are anti-seizure medications (ASMs). Other emerging pharmacologic agents to potentially treat DEE patients include serotonin modulators [e.g. EPX-100 (clemizole hydrochloride) [84, 85], EPX-200, fenfluramine [86] and lorcaserin [87]], TAK935 (soticlestat, a cholesterol 24-hydroxylase inhibitor), huperzine analogue, stiripentol (an anti-convulsant with an unknown mechanism of action) [86], ataluren (a suppressant of premature stop codons) [87], selective sodium channel modulators (e.g. verapamil) [87] and antisense oligonucleotide therapy (e.g. STK-001) [87] are in the developmental stage [88]. The first precision gene-targeted therapy specific to DEE remains challenging.
The role of the σ1R in the development of DEE is thoroughly reviewed by Martin et al. (2021) [89]. The σ1R, in the brain, acts on neuronal networks at the cellular level to fine tune neuronal signals, thus maintaining a balance between excitatory and inhibitory signals [90]. The σ1R achieves this by interacting with multiple signalling pathways and proteins and becoming activated by physiological stressors, including the excitatory/inhibitory neuronal imbalances that lead to seizure activity. In σ1R KO mice, there is a higher susceptibility to convulsive seizures caused by GABA antagonists pentylenetetrazol and (+)-bicuculline [90]. An imbalance in excitatory and inhibitory neuron signalling results in seizures. Inadequate control of glutamate signalling at the NMDAR by GABA receptors results in increased seizure activity and σ1R activation. Since there are many psychoactive σ1R ligands, there is the potential for treating DEEs [22].
1.2.3 Huntington’s Disease
Huntington’s disease (HD) is a hereditary and progressive neurological disorder caused by a Huntingtin (HTT) gene mutation. The mutation results in an abnormal form of the protein that accumulates in brain cells, leading to impairment of several neuronal signalling [91] and protein degradation [92] pathways and mitochondrial function [93], resulting in brain cell degeneration and eventual death.
The symptoms of HD usually appear in adulthood but can sometimes start in childhood or adolescence. They include uncontrolled movements, such as chorea (involuntary jerking), and cognitive and psychiatric symptoms, including impaired judgment, difficulty concentrating, depression and personality changes [94].
HD is inherited in an autosomal dominant manner, meaning that if a person has the mutated HTT gene, they have a 50% chance of passing it on to their children. There is currently no cure for Huntington’s disease, and treatment focuses on managing symptoms, such as through medication and therapy. The disease can have a significant impact on an individual’s quality of life, as well as on their family and caregivers [95].
The σ1R has been shown to play a role in HD. The exact mechanism by which the σ1R contributes to the development and progression of the disease is still being studied. There are several possible ways in which it may be involved.
One way the σ1R is thought to be involved in HD is through its interaction with the mutant huntingtin protein. In cell models of HD that express the mutated HTT protein, the σ1Rs are expressed in the nuclear inclusions and help to control the degradation of malformed proteins. Knockdown of the σ1R using small interfering RNA (siRNA) increased the volume of aggregates in the nucleus and cytoplasm and caused nuclear inclusions. Moreover, overexpression of the σ1R reduced the nuclear inclusions [96]. Studies have shown that the σ1R can bind to mutant huntingtin and may help to promote its toxic effects on brain cells. In addition, the σ1R may also be involved in regulating cellular processes disrupted in Huntington’s disease, such as protein folding and calcium signalling.
Studies have shown that expression of the σ1R is increased in the striatum of mouse models and patients with severe HD [97]. This contradicts the previous experiments; however, it may be explained by the fact that the huntingtin protein and the σ1R are involved in transporting vesicles and proteins. Therefore, overexpression of one may lead to the downregulation of the other. In the case of HD, neither protein is overexpressed, but the mutated huntingtin protein causes loss of function. As a result, the σ1R is upregulated to compensate for this loss of function.
Evidence suggests that the σ1R may be a potential target for treating Huntington’s disease. Studies have shown that drugs that activate the σ1R can help to reduce symptoms and slow the progression of the disease in animal models [98, 99]. However, more research is needed to determine whether these drugs will be effective in humans.
1.2.4 Motor Neuron Disease/Amyotrophic Lateral Sclerosis
Motor neuron disease (MND) is a progressive neurological disorder with particular damage to motor neurons responsible for controlling skeletal muscle activities involving limbs, speech, swallowing and respiratory wall muscles.
MND encompasses a spectrum of different motor neuron syndromes, including amyotrophic lateral sclerosis (ALS), progressive muscular atrophy (PMA), primary lateral sclerosis (PLS), and upper and lower motor neuron dominance [100, 101]. Among them, the most common form of MND is ALS which affects both upper and lower motor neurons and begins with limb weakness (i.e. arms, hands and legs) before progressive paralysis and death within 2–5 years after the symptom onset. The onset of MND is reported to be sporadic and varied among patients, and its pathogenesis is not yet fully understood.
A complex interaction between genetic and environmental factors is reported to trigger pathologic development and expansions. This involves glutaminergic pathways leading to glutamate-induced excitotoxicity and mutations of genes such as c9orf72, TDP-43 and fused in sarcoma (FUS), resulting in abnormalities of translation and formation of intracellular neuronal aggregates. In contrast, mutations in the superoxide dismuates-1 (SOD-1) gene increases oxidative stress [102, 103].
For treatment, palliative care is primarily advised. No effective medication is available apart from riluzole, a glutamatergic neurotransmission inhibitor, and sodium phenylbutyrate/ursodoxicoltaurine, an inhibitor of histone deacetylases (HDACs), the only drugs approved by the US Food and Drug Administration (FDA) that can delay ALS progression and improve survival by 2–3 months [104, 105].
Another drug recently approved by the US FDA, but limited to use only in Japan and the USA, is edaravone, a free radical scavenger that halts progression in early stages [106]. Although edaravone treatment has beneficial effects, its precise mechanism and cellular/molecular targets are uncertain. Masitinib, a tyrosine kinase inhibitor, has successfully passed a randomized controlled phase II/III trial in ALS patients as an additional drug for riluzole by showing a significant improvement in ALS progression, quality of life, respiratory function and time delay to death [107]. However, further trials were halted in 2021 when observational studies indicated an increased risk of ischaemic heart disease [108].
The σ1R is highly expressed in motor neurons, particularly in the ER at the postsynaptic subsurface cisternae of cholinergic C-terminals. It is hypothesized to help transport ions across voltage-gated potassium and small conductance calcium-activated potassium channels (SK) channels, thereby reducing motor neuron excitability [109, 110]. It is also speculated that higher conductance of potassium channels can also be indirectly mediated by the interaction of σ1R with the IP3R3 receptor and the subsequent amplification of calcium release [16, 111].
Consistently, reports imply that genetic mutation in the σ1R gene and the lack of functioning of the protein can result in MND [112]. The σ1RE102Q mutation (familial ALS) causes the cytoplasmic aggregation of σ1R, making them unable to bind with IP3R with loss of function to control Ca2+ flux, activated calpain and reduced ATP synthesis alongside mitochondrial-associated membrane collapse [113]. Also, studies from transgenic Drosophila expressing σ1RE102Q confirmed the same, and showed that overexpression of IP3R or glucose transporter GluT-3 could counteract σ1RE102Q toxicity [114]. Upon σ1R knockdown, the lipid raft markers, including Flotillin-1, Erlin-2 and Caveolin-1, were observed to be localized in non-raft regions, indicating the disruption of lipid rafts and associated intracellular calcium dyshomeostasis [115].
Ectopic expression of σ1RE102Q causes structural alteration in the mitochondria and ER, and induces defective autophagy, vesicular transport and mislocalization of RNA-binding proteins, leading to protein quality control modulation [116]. In ALS model mice, SOD1*G93A, knockout of σ1R exhibited deregulated motor performance, ultimately resulting in a faster death [117]. Overexpression of σ1RE102Q missense mutation in motor neuron cellular models showed high susceptibility to apoptosis triggered by endoplasmic reticulum stress [118].
Furthermore, the cells overexpressing σ1RE102Q exhibited abnormal mislocalization of the TAR DNA-binding protein (TDP-43), resulting in a toxic cytoplasmic gain of function [119, 120]. Impaired TDP-43 regulation results in unfolded protein accumulation, defective RNA metabolism and oxidative damages [121]. Misfolding or mutation in TDP-43 increases its aggregation propensity, and the intensity of motor neuron degeneration is directly correlated with TDP-43 aggregation [122]. In the cellular ALS model, σ1R stabilises nuclear pore proteins (Nups) by co-localizing with Ran-activating protein (RanGAP) and rescues nucleocytoplasmic transport deficit to reverse the ALS pathology, further extending the function and confirming the importance of the receptor [123].
1.2.5 Multiple Sclerosis
Multiple sclerosis (MS) is a neurodegenerative disease of the CNS, predominantly mediated by the autoimmune system. The myelin sheaths of neurons are permanently destroyed by immune cells causing acute inflammation and axonal transection lesions. After that, the chronic phase enters with remyelination and an unrepaired inflammation [124]. T- and B-cell infiltrates are correlated with demyelination [124]. MS is commonly found in young adults, especially women, aged between 20 and 30 years [125]. MS symptoms and the degree of severity are various, depending on the number, type and location of damaged nerves, and lead to a non-traumatic neurological disability affecting physical function, cognition, quality of life and employment. Fatigue has been reported to be a common symptom in 75% of individuals with MS [126]. Fatigue could be due to the involvement of the immune system, axonal loss, sleep disorders and circadian rhythm abnormalities during MS [127].
The treatment of MS is considered according to the disease status, whether relapsing–remitting MS (RRMS, defined as relapses at the onset with stable neurologic disability between episodes), secondary progressive MS with activity (SPMS, defined as steadily increasing neurologic disability following a relapsing course with evidence of ongoing inflammatory activity) or primary progressive MS (PPMS, defined as steadily progression from onset). Currently, there is no cure for MS, and disease-modifying therapies (DMTs) are primarily prescribed as they reduce annual relapse rates and short-term disability via modulation in immune systems, including sequestration of lymphocytes, Th1/Th2 shift, DNA synthesis in lymphocytes and cytokine production [125]. Interferon (IFN)-β-1b was the first DMT approved by the US FDA to replace the former immunosuppressants such as azathioprine, methotrexate and corticosteroids [128].
Several monoclonal antibodies (mAbs) with well-clarified mechanisms of action, including ocrelizumab, natalizumab, ofatumumab and alemtuzumab, are also approved [129]. Ocrelizumab is selective to B cells expressing CD20 and causes cell destruction and reduced inflammation. This is the only drug approved to treat PPMS. Natalizumab is specific to the α4 integrin subunit on lymphocytes and prevents lymphocytes from entering the CNS, whereas alemtuzumab selectively binds CD52 antigen on lymphocytes, leading to their depletion [129].
Besides those mAbs, sphingosine 1-phosphate receptor (S1P) modulators (e.g. fingolimod, siponimod, ponesimod and ozanimod) have been shown to bind a subtype of S1P receptors, resulting in sequestration of lymphocytes in lymph nodes [130,131,132]. Glatiramer acetate is another relatively safe DMT, even though its action is unclear [133]. Other DMTs, including fumarates (e.g. dimethyl fumarate and diroximel fumarate), teriflunomide (a pyrimidine synthesis inhibitor with anti-inflammatory and immunomodulatory activities) and cladribine (an immunomodulatory agent selectively targeting lymphocyte subtypes) are also available for treatment [134, 135].
In CNS, oligodendrocytes form myelin sheaths, including proliferation, migration and differentiation of oligodendrocyte precursor cells (OPCs). Studies have reported the co-localization of σ1R in progenitor and matured oligodendrocytes, implying a role in myelination [136]. In line with this, a study has indicated that σ1R regulated oligodendrocyte differentiation and myelin sheath generation, while σ1R siRNA showed the opposite effect [137]. Also, σ1R could interact with cholesterol, and dysfunction of the protein causes perturbations in cholesterol levels, which in turn can cause myelin degeneration [2, 138, 139]. The σ1R agonist donepezil could restore mitochondrial functions in the induced circadian rhythm disruption cellular model, suggesting the role of the receptor in maintaining a healthy circadian rhythm and possibly reducing fatigue [140]. Further, targeting σ1R reduces neuroinflammation and demyelination in the experimental autoimmune encephalomyelitis MS model, indicating the potential of the receptor to counteract the pathological effects [141].
1.2.6 Parkinson’s Disease
Parkinson’s disease (PD) is a common neurodegenerative disease with impairment in the CNS and motor symptoms. The early signs are usually tremors in one hand causing stiffness (rigid muscles) or slow movement (bradykinesia). Other symptoms can be poor balance; loss of automatic movements such as blinking, smiling and swinging arms while walking; and limitations in speech and writing.
Several biomarkers have been introduced to identify PD at the early stage because most of the cardinal symptoms occur late and dopaminergic neurons are already destroyed [142]. However, only a few have been implemented in clinical practice [143]. Neuroimaging and biochemical measurements of biofluids are the current focus due to a better prediction of PD [144].
There is currently no therapy slowing or arresting the PD progression available, so symptomatic treatment using levodopa (or in combination with carbidopa) as the first-line drug together with personalized non-pharmacological management (e.g. deep brain stimulation, strength training, gait and balance training and hydrotherapy) from a multidisciplinary team approach are primarily suggested [145, 146]. Although levodopa has been proven to improve disability and discomfort in PD patients through the conversion of l-3,4-dihydroxyphenylalanine (L-DOPA) into DA in striatum, the complete mechanism of actions and causes of its adverse events, such as dyskinesias and levodopa resistance from long-term use, are not fully understood [147, 148].α-Synuclein deposition in the brain and the formation of Lewy bodies are thought to be a hallmark of PD-related dementia characterized by changes in thinking, movement, behaviour and mood [149]. PD-related dementia and dementia with Lewy bodies are the second most common causes of dementia behind AD, accounting for 15–20% of the incidences of dementia globally [150]. Drugs targeting the build-up of Lewy bodies could potentially help prevent PD-related dementia.
There is a reduction in σ1R expression in PD, much like in AD [151], plus there is an age-related loss of dopaminergic neurons in σ1R knockout mice [152]. The σ1R may also alleviate DA toxicity in PD. High availability of DA may act as a neurotoxin, and ischaemia, hypoxia and local exposure to neurotoxins, such as high concentrations of excitatory amino acid and methamphetamine, all result in increased concentrations of DA. The DA neurotoxicity mechanism is linked to an imbalance in REDOX management. DA can spontaneously oxidize in vitro or in an enzyme-catalysed reaction in vivo to form reactive oxygen species (ROS) and DA-quinones. DA-quinones can bind to cysteine or cysteinyl residues on proteins, resulting in an imbalance of glutathione homeostasis (a key cellular antioxidant) [153]. ROS is increased in wild-type CHO and σ1R knockdown CHO cells treated with physiologically relevant DA concentrations (below 10 µM). However, apoptosis only increases in the σ1R knockdown cells [154]. Furthermore, DA-treated Neuro2A cells expressing APPSWE undergo ROS-mediated apoptosis [155], and this can be prevented using this σ1R agonist dipentylamine (this protective effect itself was reversed by σ-1 antagonist BD1047) [156].
Mutations in genes associated with PD, such as α-synuclein, DJ-1, LRRK-2 (which encodes leucine-rich repeat kinase 2) and GBA (which encodes glucocerebrosidase) have been identified, which cause abnormal α-synuclein proteostasis leading to mitochondrial dysfunction, oxidative stress, neuroinflammation and motor circuit pathophysiology [157].
Oxidative damage to the substantia nigra has been demonstrated in patients with PD [158]. Oxidative stress accelerates α-synuclein aggregation and cell death [159, 160]. Multiple studies have identified σ1R ligands to reduce oxidative stress through multiple in vivo and in vitro models via various signalling pathways involving protecting the ER and mitochondria [161,162,163,164,165].
Similarly to Aβ in AD, α-synuclein oligomers bind to PGRMC1 at synaptosomes in the brain [166]. Furthermore, in chaperone-controlled autophagy, α-synuclein binds to lysosome-associated membrane protein 2A (LAMP-2A) [167, 168] (LAMP-2A is the receptor for chaperone-mediated autophagy). α-Synuclein disrupts cholesterol-rich lipid membranes where the σ2R is located and disrupts downstream signalling events, including autophagy [169,170,171]. Without efficient autophagy, neurons gather ubiquitinated protein aggregates (proteins tagged with ubiquitin for degradation). Thus, allosteric antagonists of the σ2R may prevent the effects of α-synuclein oligomers via various mechanisms (possibly similar to AD and Aβ accumulation at the synaptosomes).
A high-throughput screening assay making use of the National Institutes of Health (NIH) small molecule repository (725 compounds that have been tested in phase I/III clinical trials) and the CogRx library of high-affinity σ2R allosteric antagonists, investigated the effects of these compounds on lipid vesicle trafficking deficits caused by α-synuclein [172]. The study found that the most effective compounds were the σ2R allosteric antagonists [172]. Furthermore, an α-synuclein oligomer-induced increase in LAMP-2A expression was blocked by the σ2R allosteric antagonists.
1.2.7 Rett Syndrome
Rett syndrome (RS) is a rare genetic neurodevelopmental disorder affecting brain development and consequently developing into physical, cognitive and intellectual disabilities. RS is mostly found in females [173]. The most precise cause of RS is demonstrated to be the loss-of-function mutation of methyl CpG binding protein 2 (MECP2) gene located at the X chromosome Xq28 [174].
Dysfunction of MeCP2, an epigenetic modulator of gene expression acting as both a transcriptional activator and repressor, induces either positive or negative changes in many other gene expressions at transcriptional and post-transcriptional levels [175, 176]. MeCP2 deficiency also impacts successive stages of brain development and contributes to mitochondria dysfunction [177], impaired protein synthesis [178] and reduced synaptogenesis [179], leading to pathology of RS. The important symptoms for differential diagnoses are cerebral palsy, autism, Angelman syndrome and non-specific developmental delay [173]. Spectrums of these symptoms, together with progression and severity, are also broad and individualized [180].
There is no cure available [181], so medical care and management aim to provide symptomatic relief through pharmacological and non-pharmacological approaches. Medication is needed for breathing and motor difficulties. Since over half of RS patients have seizures, anti-convulsant/anti-epileptic drugs such as valproate, lamotrigine, levetiracetam and carbamazepine are usually prescribed [182]. Serotonin-based therapy, such as serotonin 1A receptor agonists and serotonin reuptake inhibitors (SSRIs) are also reported to help reduce symptoms of sleep apnoea [183].
A wide variety of neurologic impairments characterizes RS. Unlike some neurodegenerative diseases (PD and AD in particular), there appears to be little or no causal link established between the σ1R and the progression of RS. However, due to the promiscuous relationship of the σ1R with multiple proteins and signalling pathways in the brain and its relative success as a target for treating other neurodegenerative diseases, the σ1R is a target for treating the neurological damage seen in RS.
Most patients carry a mutation in the MeCP2 gene [184]. MeCP2 is found at high levels in neurons and is associated with the maturation and synaptic plasticity of the CNS [185, 186]. MeCP2 is largely thought to negatively regulate gene expression (although there is some evidence that it activates some genes).
However, when mutated in Rett syndrome, MeCP2 target genes that would normally be activated remain inactive, and genes that would be inactivated remain active, thus producing unneeded proteins. Brain-derived neurotrophic factor (BDNF) is one protein that MeCP2 is thought to negatively regulate [187], along with the mTOR/AKT signalling pathways [178]. The σ1R is known to have a role in regulating BDNF expression in multiple cellular systems, including the brain [188], eyes [189] and heart [190]. Furthermore, many studies have identified that σ1R agonist treatment of various cells results in AKT phosphorylation [191]. Also, σ1R activity has a role in autophagy regulation, becoming a novel target for treating neurological disease [53, 65, 192].
1.2.8 Wolfram Syndrome
Wolfram syndrome (WS), also known as diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD), is a rare genetic disorder affecting many body parts. It is caused by mutations in the WFS1 gene, which provides instructions for making a protein called wolframin [193].
The most common features of the disorder include diabetes mellitus (a condition that affects how the body uses and stores glucose), diabetes insipidus (a disorder that causes excessive thirst and urination), optic atrophy (damage to the optic nerve that can cause vision loss) and deafness. WS may also include neurological problems such as ataxia (difficulty with coordination and balance), seizures, behavioural problems, urinary tract problems and psychiatric disorders.
There is no cure for WS, and treatment is based on the individual’s symptoms. Management may involve regularly monitoring blood sugar levels, hormone replacement therapy and other supportive measures such as hearing aids and visual aids [194].
Wolframin protein is expressed at the ER, interacts with the MAM and is involved in calcium transfer between the ER and the mitochondria. The mutated form of wolframin disrupts calcium transfer, leading to mitochondrial dysfunction and cell death [195]. The σ1R is heavily involved in chaperoning proteins, such as the IP3R, in calcium shuttling between the ER and mitochondria. σ1R activation, either during cellular stress or with an agonist, results in calcium transfer from the ER to the mitochondria reducing ER stress, protecting the mitochondria and improving cellular ATP levels and the general health of the cell [196]. In mouse and zebrafish models of WS, overexpression of the σ1R alleviated the symptoms and restored calcium homeostasis in vivo and in vitro [197].
2 Sigma Receptor Ligands for Neurological Disease
2.1 Amantadine
Amantadine (Fig. 2) is a weak NMDA antagonist that increases DA release and blocks DA reuptake [198, 199]. Amantadine also has a reported affinity for the σ1R of 7 µM ([3H]-(+)-SKF-10,047 in rat cerebellum or forebrain homogenates [200]) or 740 µM ([3H]-(+)-pentazocine in permeabilized MDA-MB-468 breast cancer cells [2, 3]), and also has an affinity for the σ2R of 9 µM ([3H]-DTG in membranes prepared from MCF7 breast cancer cells [5, 201]). Amantadine was previously used to treat influenza-A in the early 1960s and 1970s, although it is no longer recommended due to widespread resistance [202]. Amantadine has a mild side effect profile. The common neurological side effects include drowsiness, lightheadedness, dizziness and confusion [203].
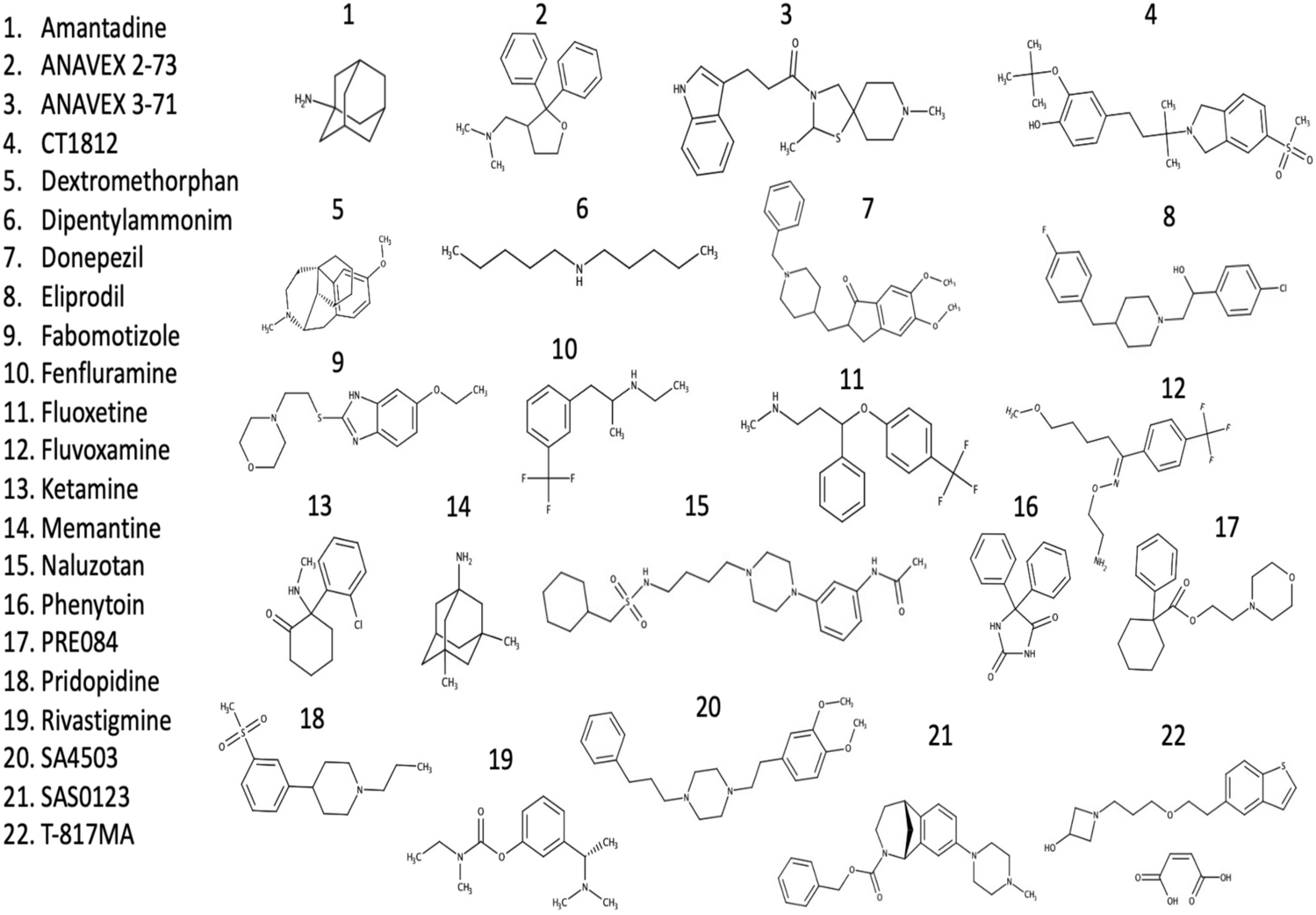
Chemical structures of drugs with sigma receptor activity discussed within this manuscript.
2.1.1 Parkinson’s Disease
Amantadine is a drug long used in treating PD [204] and has been used for treating movement disorders in PD for over 50 years. Despite its long use in treating PD, the pharmacological activity of amantadine is still not fully understood. Its use in PD has declined since the introduction of L-DOPA treatment. However, it is still used in 10% of patients today, alone or in combination with L-DOPA [205].
The blocking of [3H]-(+)-SKF-10,047 binding by amantadine in vivo in various mouse brain structures correlated with the data from the rat cerebellum or forebrain homogenates [200], and in post-mortem studies of human brains [206], suggesting a physiologically relevant interaction of amantadine with the σ1R.
The use of amantadine in treating PD and L-DOPA-induced dyskinesia has been debated, with one systematic review suggesting there is no evidence for its safety and benefit in treating PD [207]. This may be due to the dose used and the pharmacokinetics of amantadine. As plasma concentrations of amantadine increase, there is a greater risk for toxicity, particularly in patients with renal diseases (the kidneys remove most amantadine) [208]. New formulations of amantadine that release over a longer period have been trialled and approved for PD in the USA [209,210,211,212] (Table 1). The extended-release formulation is typically used to help manage PD symptoms throughout the day rather than providing short-term relief. Slowly releasing the medication over several hours can help control motor symptoms such as tremors, stiffness and difficulty with movement during both dyskinesia and OFF episodes. Immediate-release amantadine is typically taken two to four times a day, and its effects can wear off relatively quickly, which may result in fluctuations in symptom control [209]. On the other hand, extended-release amantadine is designed to slowly release the medication over several hours, providing a more stable blood level of the medication and, therefore, more stable symptom control.
| Study title | Conditions studied | Primary outcome measures | Study results | Trial Phase | Trial number |
|---|---|---|---|---|---|
| E-R amantadine safety and efficacy study in LID (EASED Study) | DyskinesiaLIDPD | Change in UDysRS total score from baseline to week 8 | E-R amantadine appeared relatively safe with minimal adverse effects at doses of 260 and 340 mg. There was also a statistically significant reduction in LID (measured using the change in UDysRS) total score. Amantadine reduced LID compared with placebo | 2/3 | NCT01397422 |
| Efficacy and safety study of ADS-5102 in PD patients with LID (EASE LID 3) | DyskinesiaLIDPD | Change in UDysRS total score from baseline to week 12 | A statistically significant improvement was measured using UDysRS total score for 340 mg amantadine compared with placebo [210,211,212, 214]. | 3 | NCT02274766 |
| ADS-5102 for the treatment of LID (EASE LID Study) | DyskinesiaLIDPD | Change in UDysRS total score from baseline to week 12 | A statistically significant change in UDysRS of −15.9 (1.6) for ADS-5102. A statistically significant change in OFF time, which decreased by a mean (SE) of 0.6 (0.3) hours for ADS-5102 and increased by 0.3 (0.3) hours for placebo [214]. | 3 | NCT02136914 |
| Open-Label safety study of ADS-5102 in PD patients with LID | DyskinesiaLIDPD | Number of participants with reported AEs and safety-related study drug discontinuations | Patients switching from IR to E-R amantadine experienced an improvement in motor complications, without exacerbating adverse events. Plus a statistically significant reduction in their MDS‐UPDRS Part IV scores at the first post‐baseline visit (week 8) plus many patients were able to reduce their levodopa use [212]. | 3 | NCT02202551 |
Clinical trials of extended-release amantadine
AEs adverse events, E-R extended release, IR immediate release LID levodopa-induced dyskinesia, PD Parkinson’s disease, SE standard error, UDysRSUnified Dyskinesia Rating Scale
Extended-release amantadine may also have a lower incidence of certain side effects, such as nausea and dizziness, compared with immediate-release amantadine. This is because extended-release amantadine releases the medication more slowly, which can reduce the peak concentration of the drug in the blood and potentially minimize side effects [213,214,215].
2.1.2 Multiple Sclerosis
Amantadine treatment improved fatigability associated with MS in several randomized controlled studies [216,217,218,219]. Amantadine treatment has been earlier reported to induce sleep spindles in PD individuals, implying the role in rectifying sleep disorders and associated fatigue during MS [220]. Amantadine has been trialled to relieve fatigue in MS patients (NCT03185065). Using a randomized cross-over study design, 100 mg of amantadine (increasing to 200 mg if tolerated) was given for 6 weeks per treatment period, followed by a 2-week washout. Fatigue (the primary endpoint) was measured using the modified fatigue impact scale (MFIS) between days 26 and 35. However, the study showed no significant improvement over the placebo and increased adverse events [221]. There have been three other clinical trials for MS with extended-release formulations of amantadine concerning drug safety and walking impairment (NCT02471222, NCT03567057 and NCT03436199). Amantadine extended release at 137 mg, (165 patients) and 274 mg (132 patients) improved walking speed in the Timed 25 Foot Walk compared with the placebo (174 patients) (NCT03436199). Furthermore, these improvements were also seen in the open-label follow-up study (NCT03567057). The data from NCT02471222 have not been made available.
2.2 ANAVEX2-73 (Blarcamesine)
Blarcamesine (ANAVEX2-73; Fig. 2) is a σ1R receptor agonist with an affinity of 860 nM (IC50) [222] developed for treating AD. Blarcamesine also has agonist activity at the muscarinic M1 receptor, and the NMDA receptor with affinities of 5 and 8 µM, respectively [222].
2.2.1 Alzheimer’s Disease
Pre-clinical studies have shown that blarcamesine can reverse scopolamine’s long-term amnesic effects in mice [222]. Furthermore, blarcamesine can reverse the effect of learning deficits in mice injected with Aβ25–35 [222,223,224]. Blarcamesine given to mice injected with Aβ25–35 restored respiration rates in hippocampal mitochondria and reduced lipid peroxidation levels. Blarcamesine could also prevent tau-hyperphosphorylation and amyloid-β (1–42) generation in mouse models of AD [223]. Furthermore, indicators of increased toxicity, including Bax/Bcl-2 ratio and cytochrome C release into the cytosol, were reduced [224]. Blarcamesine has protective effects in Caenorhabditis elegans and cell culture models of AD by enhancing autophagic flux and increasing proteostasis [65].
A phase 1 clinical study in healthy male volunteers showed that doses up to 50 mg were well tolerated, with no serious adverse events reported in a poster at the CNS summit in 2014 [225]. At 60 mg, three out of four subjects presented dose-limiting CNS symptoms (dizziness and headache). The study concluded with pharmacokinetic analysis showing extensive biotransformation of blarcamesine to AV19-144 (the bioactive form of the drug) with assumed linear pharmacokinetics after single oral dosing of 1 to 60 mg blarcamesine [225].
A biomarker analysis, phase 2a study (NCT02244541), that include some exploratory study into the benefits of blarcamesine, where blarcamesine was administered over 57 weeks, showed a significant reduction in cognitive decline [226]. When the study was extended to 265 weeks (NCT02756858), there was confirmation of this prevention of decline at 148 weeks with analysis of blood concentrations of blarcamesine and its metabolite AV19-144, showing that those with high concentrations of blarcamesine had improved therapeutic responses of 78% and 88% in adjusted MMSE and adjusted AD cooperative study–activities of daily living (ADCS–ADL), respectively, relative to the low/medium concentrations of blarcamesine.
Furthermore, genomic analysis also indicated the significant effects of biomarkers on clinical outcomes of blarcamesine. Two patients showed an exceptional therapeutic response at 148 weeks. Both subjects had σ1R wild type and a high mean concentration of blarcamesine in plasma, with a baseline MMSE > 20 [226].
The study of biomarkers that predict the potential therapeutic outcome of a drug is especially useful in a disease with such heterogeneity as AD [226]. The ApoE ε4 genotype alone lacks predictive power since only approximately 25% of people carry this genotype. The study discussed above identified that patients with a baseline MMSE greater than 20 who had the σ1R wild type with a high mean blarcamesine serum concentration and ApoE3 alleles had significantly better outcomes in the ADCS–ADL. The SIGMAR1 Q2P variant, having a baseline MMSE score lower than 20 and a low mean serum concentration of blarcamesine were predictive of poor outcomes in the ADCS–ADL [226].
Blarcamesine is undergoing further phase 2b/3 clinical trials (ongoing: NCT04314934; completed with data yet to be published: NCT03790709). The results of these clinical trials will be interesting, given the promise blarcamesine has shown in the pre-clinical studies.
2.2.2 Multiple Sclerosis
Blarcamesine increased the proliferation of oligodendrocyte progenitor cells to myelin sheath-producing oligodendrocytes in a σ1R-dependent manner and protected them from excitotoxic insults [227]. There are no currently listed clinical trials for blarcamesine in MS on www.clinicaltrials.gov.
2.2.3 Rett Syndrome
Long-term administration of blarcamesine to mice deficient in MeCP2 restores physiological and neurological abnormalities that mimic the human Rett syndrome, including ameliorating motor and acoustic startle deficits and reversing expiratory apnoea [228]. Several σ1R-related mechanisms could benefit the neurological and motor symptoms seen in this and other models of Rett syndrome. Enhancement of calcium homeostasis via σ1R shuttling of calcium to the mitochondria leads to improved mitochondrial function and the activation of autophagy via σ1R activation, resulting in a reduction of misfolded proteins and fewer damaged organelles or the restoring/upregulation of BDNF [65, 228, 229]. This leads to potential improvements in synaptic function in multiple brain regions.
Blarcamesine is currently in phase 2/3 clinical trials (NCT03758924, NCT03941444 and NCT04304482). Blarcamesine has shown positive results in a phase 2 clinical trial (NCT03758924) in female patients diagnosed with Rett syndrome (positive MeCP2 gene mutation), meeting both primary and secondary endpoints with no reported serious adverse events (press release) [230]. In another phase 3 (NCT03941444) clinical trial the primary and secondary endpoints were met with significant improvements versus placebo in the clinical global-impression of improvement scale (CGI-I); the anxiety, depression and mood scale (ADAMS); and drug exposure-dependent response of the Rett Syndrome Behaviour Questionnaire (RSBQ), all with a low incidence of adverse events (press release) [230]. It is important to note that the data available from these studies has not been peer reviewed and formally published. There are further ongoing phase 3 studies organized by Anavex Life Sciences (NCT04304482). However, while the data look promising, it will be important to see some independent studies with peer-reviewed results to fully assess the efficacy and safety of blarcamesine for Rett syndrome treatment.
2.3 ANAVEX3-71
ANAVEX3-71 (AF710B) (Fig. 2) is an agonist at the muscarinic M1 receptor and σ1R, and the second drug developed by for AD treatment. ANAVEX3-71 has an affinity for the σ1R of 1.3 nM and an affinity for the σ2R of 10 µM [62]. The affinity for the M1 receptor is approximately 0.05 nM. ANAVEX3-71 also has affinities for the µ opioid receptor and serotonin receptor (approximately 10 µM for each) [62].
2.3.1 Alzheimer’s Disease
Given the complex pathology of AD, it is of interest that a drug such as AF710B may interact with multiple target sites to have a beneficial effect in AD [52].
Pre-clinical studies have shown ANAVEX3-71 to be worthy of further study. AF710B has been shown to modify the disease-defining hallmarks of AD in transgenic (3×Tg-AD) mice, such as cognitive deficits, amyloid and tau pathologies, and beneficial effects on mitochondrial dysfunction and neuroinflammation [62, 231].
Phase 1 clinical trials (NCT04442945) in healthy individuals have been reported on Anavex Life Sciences’ website, suggesting that in healthy volunteers, 5–200 mg daily had no serious adverse events. The pharmacokinetics of ANAVEX3-71 (serum concentration of AF710B) was also proportional to the dose for doses up to 160 mg. Furthermore, there were no clinically significant electrocardiogram (ECG) parameters throughout the study (press release) [232].
There are no further registered (www.clinicaltrials.gov) ongoing clinical trials with ANAVEX3-71. However, Anavex states on its website that they plan further phase 2 clinical trials. More independent studies are required. However, pre-clinical data on ANAVEX3-71 suggest it could be useful in treating dementia and AD.
2.4 CogTX Compounds
CT1812 (Fig. 2) is one of several σ2R antagonist compounds developed by Cognitive Therapeutics Inc. (CogTx) for treating AD [233]. Other similar σ2R compounds from this company that have a strong affinity for the σ2R are CT1978 (Kd = 9.2 nM), CT1970 (Kd = 1.8 nM), CT2168 (Kd = 2 nM), CT1681 (Kd = 12 nM), CT1696 (Kd = 11 nM) and CT1925 (Kd = 18 nM), and act as allosteric σ2R antagonists [172].
CT1812 has become the company’s lead compound, with higher selectivity of the σ2R and improved metabolic stability. Compounds CT01346 and CT01344 had a low polar surface area and off-target effects at the Ether-a-go-go-related gene (ERG) and at other sites. CT1812 has an affinity of 8.5 nM for the σ2R [233].
2.4.1 Alzheimer’s Disease
CT1812 has been demonstrated in rats to prevent Aβ-oligomer binding to neurons and to displace bound Aβ-oligomers from neuronal receptors [234, 235]. CT1812 does not block oligomer formation, suggesting that its ability to stop Aβ-oligomer binding and trafficking deficits are a result of σ2R binding. Aβ oligomer concentrations were measured in the brain interstitial fluid (ISF) of 12-month-old APPSwe/PS1dE9 transgenic mice using electrodes coated with the oligomer-specific antibody placed in the hippocampus. CT1812 resulted in a rapid and significant increase in Aβ-oligomer levels (not seen with the vehicle control) but did not affect Aβ monomer levels.
Furthermore, post-mortem tissue samples observed a similar displacement of Aβ-oligomers after treatment with CT1812 [175]. CT1812 is the first drug for AD to work by displacing Aβ-oligomer from synaptic receptors and being the first σ2R antagonist. Interestingly, CT1812 mimics the protective effects of the Icelandic A673T mutation in the APP gene, which may be predictive of clinical success [236].
CT1812 has been studied in phase 1 clinical trials and is relatively safe (NCT03716427 and NCT02907567) at doses up to 560 mg, resulting in minimal adverse events in healthy volunteers (reported in an abstract from the Alzheimer’s Association) [237, 238]. In a placebo-controlled biomarker study (NCT02907567) in mild AD patients treated with CT1812, there was a significant increase in CSF Aβ-oligomer concentrations and decreased synaptic protein fragments neurogranin and synaptotagmin relative to baseline compared with placebo-treated patients [235]. There are several phase II clinical trials, either ongoing (SEQUEL study, NCT04735536, SHINE study, NCT03507790, NCT05225415 and NCT05531656) or completed, without published results or results made available (SNAP study: NCT03522129, NCT05225389, NCT05248672 and NCT02570997). The SPARC study (NCT03493282) aimed to assess the effects of CT1812 on synaptic density and suggests an increase in synaptic density based on the available data found at www.clinicaltrials.gov, along with imaging results and statistical analysis, which have yet to be formally published. In the same study, results posted online appear to show a reduction in CSF Aβ40, CSF Aβ42, CSF tau, CSF phospho-tau, CSF neurogranin (NRGN), CSF(SNAP25) and CSF, and increases in CSF synaptotagmin and neurofilament light (NFL). However, there appeared to be no obvious improvements in the cognition scores, MMSE or the AD Assessment Scale–Cognitive Subscale (ADAS–COG), although without complete data analysis and statistical study, it is difficult to comment on these study aspects.
2.4.2 Parkinson’s Disease
α-Synuclein binds to LAMP-2A, and mutated forms bind to LAMP-2A tighter, preventing degradation of other substrates and inhibiting the autophagic process. The CogTx compounds were screened in primary neuronal cells against α-synuclein oligomers. Each compound inhibited α-synuclein oligomer-induced lipid vesicle trafficking deficits in a dose-dependent manner [CT1978 (EC50 = 1.48 μM), CT1970 (EC50 = 6.50 μM), CT2168 (EC50 = 1.5 μM), CT1681 (EC50 = 9.44 μM), CT1696 (EC50 = 15.53 μM) and CT1925 (EC50 = 8.95 μM)]. Two more potent compounds, CT1978 and CT2168, were also tested against LAMP-2A expression, and both prevented α-synuclein-induced upregulation of LAMP-2A [172]. Taken together, the data from this study suggest that σ2R ligands (antagonists) may potentially regulate chaperone-mediated (LAMP-2A) autophagy and have a potential role in PD therapy.
The CogTx σ2R compound CT1812 discussed above regarding AD is also in phase 2 clinical trials to assess the efficacy of CT1812 in patients with Lewy body dementia (NCT05225415).
No other studies/trials are registered at www.clinicaltrials.gov regarding the above compounds or any other σ2R ligand. However, these compounds appear to have potential in treating PD, and their activities through the σ2R warrant studying other σ2R antagonists in relation to PD.
2.5 Dextromethorphan
Dextromethorphan (DXM) (Fig. 2) is a drug used in cough-suppressing medicines and targets multiple receptors, including inhibiting serotonin reuptake [239] and acting as an agonist at the σ1R [240]. DXM has an affinity for the σ1R of between 142 and 652 nM, and between 2 and 9 µM for the NMDA receptor [241]. DXM is metabolized into its active metabolite dextrorphan (DXO) by the enzyme cytochrome P450 (CYP)2D6, a much more potent NMDA receptor antagonist (400–900 nM) but a much less potent serotonin reuptake inhibitor than (400–800 nM) DXM (20–40 nM) [240]. Dextromethorphan/quinidine (AVP-923; Nuedexta) is developed and marketed by Avanir Pharmaceuticals. Adding quinidine to the DXM reduces its metabolism to DXO by inhibiting CYP2D6. Deuteration of DXM (AVP-786, also produced by Avanir Pharmaceuticals) further hinders its metabolism by CYP2D6, allowing for lower doses of quinidine in the combination. AXS-05 combines DXM and the atypical antidepressant drug bupropion (developed and marketed by Axsome Therapeutics). In this combination, bupropion also acts as a potent CYP2D6 inhibitor, preventing the bioactivation of DXM.
2.5.1 Alzheimer’s Disease
Pseudobulbar affect (PBA) is a condition linked to various neurological disorders and manifests as uncontrollable laughing or crying. There is evidence to suggest that σ1R agonists act to reduce PBA in patients with neurological diseases. The mechanism by which DXM is thought to treat PBA is related to NMDA and σ1R binding; it is also noted that dextromethorphan most prominently binds in the brainstem and cerebellum, areas particularly rich in σ1R and is implicated in PBA [242]. DXM, with the addition of quinidine inhibiting its metabolism, has been shown to positively affect the symptoms of PBA [243]. Dextromethorphan/quinidine is the first FDA-approved drug for the treatment of PBA.
Agitation is another symptom of AD and is one of the most distressing for caregivers, resulting in institutionalization, further isolation and reduced quality of life for the AD patient. There have been multiple studies investigating DXM in combination with other drugs (AVP-923 and AXS-05) or the deuterated form of DXM in combination with quinidine (AVP-786) [244,245,246]. The registered trials with DXM and DXM-related drug combinations are listed in Table 2.
| Study title | Conditions studied | Primary outcome measures | Study results | Trial phase | Trial number |
|---|---|---|---|---|---|
| Efficacy, safety and tolerability study of AVP-923 (dextromethorphan/quinidine) for treatment of symptoms of agitation in participants with Alzheimer’s disease | Agitation in AD | Change in the NIP Agitation/Aggression Domain Score from day 1 to day 36 and from day 36 to day 70 | A significantly reduced NPI Agitation/Aggression scores for dextromethorphan-quinidine versus placebo [244] | 2 | NCT01584440 |
| Nuedexta in the treatment of pseudobulbar affect in patients with Alzheimer’s disease | PBA in AD | Reduction of PBA frequency | No results posted (last update 2019) | 4 | NCT01832350 |
| Open-label safety study of AXS-05 in subjects with Alzheimer’s disease agitation | Agitation in AD | Incidence of treatment-emergent adverse events following dosing with AXS-05 | Study ongoing (last update 2023) | 3 | NCT04947553 |
| A study to assess the efficacy and safety of AXS-05 in subjects with Alzheimer’s disease agitation | Agitation in AD | Cohen–Mansfield Agitation Inventory (CMAI) | AXS-05 significantly relieved agitation, a mean percentage reduction from baseline of 48% for AXS-05 versus 38% for placebo [246] | 3 | NCT05557409 |
| Assessing clinical outcomes in Alzheimer’s disease agitation (AXS-05) | Agitation in AD | Time from randomization to relapse of agitation symptoms | Study ongoing (last update 2022) | 3 | NCT04797715 |
| Long-term extension study of the safety and efficacy of AVP-786 for the treatment of agitation in patients with dementia of the Alzheimer’s type | Agitation in AD | Number of participants with any type of adverse event. Sheehan Suicidality Tracking Scale, Mini-Mental State Examination and Epworth Sleepiness Scale | Study ongoing (last update 2023) | 3 | NCT02446132 |
| Efficacy, safety and tolerability of AVP-786 for the treatment of agitation in patients with dementia of the Alzheimer’s type | Agitation in AD | Change from baseline in the CMAI Composite Score to week 6 and week 12 | AVP-786 showed some statistically significant reductions in agitation measured by CMAI and mADCS-CGIC-Agitation score | 3 | NCT02442765 |
| Efficacy, safety and tolerability of AVP-786 for the treatment of agitation in participants with dementia of the Alzheimer’s type | Agitation in AD | Change from baseline to week 12 in the CMAI Composite Score | AVP-786 showed some statistically significant improvement versus placebo in the secondary outcome in change from baseline to week 12 in the NPI Agitation/Aggression Domain score | 3 | NCT02442778 |
| Comparative effectiveness of MCI and dementia treatments in a community-based dementia practice | Mild cognitive impairment | Cognitive testing – Memory Orientation Screening Test (MOST) | No results published (last update 2016) | 3 | NCT02860338 |
| Safety study of AVP-923 in the treatment of IEED also known as PBA | PBA in AD | Emotional control | No results published (last update 2016) | 3 | NCT00056524 |
Clinical trials of dextromethorphan, and drug formulations of dextromethorphan for AD and AD-related symptoms.
AD Alzheimer’s disease, CIMA Cohen–Mansfield Agitation Inventory, mADCS-CGIC Mean Modified Alzheimer’s Disease Cooperative Study–Clinical Global Impression of Change, IEED Involuntary Emotional Expression Disorder, NIP Neuropsychiatric Inventory, PBA pseudobulbar affect.
DXM has an affinity of 205 nM for the σ1R, whereas its metabolites DN and dimemorfan (DF) have affinities of 151 and 144 nM, respectively. All three compounds have a low affinity for the σ2R (4–11 μM). DN has a higher affinity for the PCP site of the NDMAR (900 nM), whereas DXM and DF have affinities of 7 and 17 µM, respectively [247].
2.5.2 Parkinson’s Disease
Intraperitoneal injections (IP) of DXM (20 mg/kg) to 6-OHDA-treated rats significantly attenuated 6-OHDA-induced DAT and SERT loss, correlating to rotational behaviours [4]. DXM also has anti-dyskinetic activity [248, 249]. For a long time, it was assumed that these effects were mediated via the NMDA receptor since DXM has moderate to low affinity at this receptor and acts as an antagonist. However, it has been confirmed that the σ1R is responsible (at least in part) for the anti-dyskinetic effects since other NMDA antagonists, MK-801 and HA-966, failed to suppress dyskinesias in the 6-OHDA rat model, whereas DXM did and the σ1R antagonist BMY-14802 could reverse this [250].
Analysis of patient data between January 2005 and December 2013 showed that low cumulative doses of DXM may potentially achieve a protective effect for PD; however, high cumulative doses seem to be a risk for PD [251].
DXM/quinidine 45 mg/10 mg twice daily has been clinically trialled (NCT01767129) to treat levodopa-induced dyskinesia in PD. The small study provided evidence of the clinical benefit of the drug 10 mg twice daily (BID) for treating L-DOPA-induced dyskinesia in patients with PD [252]. These results aligned with two older studies showing similar anti-dyskinetic effects of DXM alone [249, 253]. However, at the time of writing, DXM (and its formulations) have only been approved for the treatment of PBA and uses in other conditions have been discontinued.
In a case study of a patient with Lewy body dementia who exhibited agitation and aggression (common behavioural and psychological symptoms of Lewy body dementia), the patient was prescribed an acetylcholinesterase inhibitor (rivastigmine) in combination with different anti-psychotics, antidepressants with anti-histaminic effects, anti-convulsants and benzodiazepines, without effect on the outbursts of aggression (paradoxically, the agitation and aggression worsened). The caregiving physician then tried DXM in combination with the CYP2D6 enzyme-inhibiting drug paroxetine. Within 1 day, the patient’s behaviour improved with the cessation of the aggression. The patient remained on the DXM/paroxetine combination plus the anti-psychotic drugs. However, 6 weeks post-discharge from the hospital, the patient committed suicide. It is unclear whether any prescribed drugs had a causal relationship with the patient taking their life [254]. However, there is reported evidence that DMX abuse can lead to assault, suicide or homicide [255].
2.5.3 Motor Neuron Disease/Amyotrophic Lateral Sclerosis
As the first FDA-approved drug for the treatment of PBA, dextromethorphan/quinidine enhanced the bulbar function (speech, swallowing and salivation) in ALS individuals in a randomized clinical trial [256, 257], and several other trials have been completed (NCT01806857, NCT03883581, NCT01806857 and NCT00573443). However, in the long term, the drug does not affect the deterioration of bulbar function or avert non-invasive respiratory management in ALS individuals [258].
2.5.4 Multiple Sclerosis
Long-term treatment with DXM in experimental animal models of MS showed a reduction in neutrophil infiltration through the inhibition of NOX-2 and attenuated axonal demyelination through the upregulation of σ1R [259]. In addition, DXM stimulated the proliferation and maturation of oligodendroglia progenitor cells to oligodendrocytes and protected against excitotoxicity, indicating neuroprotective effects [260].
Two placebo-controlled trials with dextromethorphan/quinidine have shown an effective therapy for PBA in MS patients [261, 262], and treatment with fluoxetine and DXM also produced a similar effect in women with MS [263].
A common symptom of MS is chronic pain [264]. The σ1R has long been associated with pain since its identification and misidentification as a novel opioid receptor [265]. Panitch et al. [266] showed evidence supporting the off-label use of dextromethorphan/quinidine for pain reduction in MS. However, pain measurement was not a primary outcome measure for those particular trials. DXM has been trialled for treating central neuropathic pain in multiple sclerosis (NCT01324232). There have been at least five studies into the use of DXM in pain treatment from various conditions other than MS, with the results pointing towards a possible pain-relieving effect of DXM (unrelated to MS) [266,267,268,269,270,271]. Concerning the σ1R, pain-relieving drugs studied tend to be antagonists [40], and DXM is a σ1R agonist. Thus, any pain-relieving effect may not be due to DXM’s activation of the σ1R.
2.5.5 Rett Syndrome
DXM has also been trialled in Rett syndrome patients (NCT00593957, NCT01520363, NCT00069550 and NCT00004807). Two trials have published results at www.clinicaltrials.gov (NCT01520363 and NCT00593957) [272]. In the two groups (PCTDMRTT–NCT01520363), 24 patients in DXM (5 mg/kg per day) and 26 in placebo, were evaluated in the Mullen Scales of Early Learning (Mullen) at the end of the three-month treatment period. The open-label study had three groups, each receiving either 0.25, 2.5 or 5 mg/kg/day. At six months, the improvement in receptive language was measured by the Mullen Scale and the Screen for Social Interaction (SSI) score (mean and difference from baseline) were recorded. The data from these studies do not look promising. While the safety of the drug was not an issue, with only a few minor adverse events, the study showed little efficacy in the primary and secondary outcomes in both studies.
2.6 Dipentylammonium
Dipentylammonium (DPA) (Fig. 2) is an N-alkyl-amine similar in structure to that of (2R-trans)-2-butyl-5-hetylpyrrolidine (σ1R Ki 2nM [273]), tridemorph (σ1R Ki 0.3–30 nM [274]), stearylamine (σ1R Ki 8 µM [275]), dodecylamine (σ1R Ki 183 nM [274]) and endogenous sphingolipids [276]. DPA has an affinity for the σ1R (Ki 17 nM), and it binds to the same binding site as (+)-pentazocine in a competitive fashion [2, 5, 156].
2.6.1 Alzheimer’s Disease
DPA has been shown to prevent oxidative stress and ER stress in HT-22 cells exposed to 5 mM glutamate. Reducing cell death and ROS production in a σ1R antagonist sensitive manner. Furthermore, DPA increased neurite outgrowth in Neuro2A cells and prevented DA toxicity in APP(Swe) overexpressing Neuro2A cells [156].
There are no clinical safety or efficacy data for DPA, and its simple structure will likely hinder potential clinical studies due to concerns about its specificity. DPA, however, is particularly selective for the σ1R, over the σ2R, monoamine transporters, DA receptors and muscarinic AchR [2].
2.7 Donepezil
Donepezil (Aricept) (Fig. 2) is one of the current standard acetylcholinesterase inhibitors (AChEI) that are prescribed for mild-to-moderate AD, along with galantamine (Reminyl), rivastigmine (Exelon) and memantine (Ebixa), according to the online guidance from the National Institute for Health and Care Excellence (NICE) in the UK [276]. Donepezil appears to provide improved cognition in mild-to-moderate AD, although it does not halt the progression of the disease. Donepezil has an affinity for the σ1R (Ki 14.6 nM) [277], and studies using rats have shown that there is 90% occupancy of σ1Rs in the brain when dosed with 10 mg/kg [278].
2.7.1 Alzheimer’s Disease
Donepezil has been associated with reduced cardiovascular mortality in patients with AD [279, 280], likely due to cardioprotective effects of σ1R ligands [281]. Studies investigating donepezil’s σ1R targeting activities have shown that it can potentiate nerve growth factor (NGF)-induced neurite outgrowth in PC12 cells [282] (a common test for σ1R agonist activity). Furthermore, positron emission tomography (PET) scans in mice have shown that donepezil can displace 11C-SA4503 from the brain dose dependently at physiologically relevant doses [278].
Donepezil has been thoroughly tested clinically in moderate-to-severe AD, with many studies (discussed in multiple meta-analyses and systematic reviews [283,284,285]) indicating a statistically significant improvement in the standardized Mini-Mental State Examination (SMMSE, on which scores range from 0 to 30, with higher scores indicating better cognitive function) and the Bristol Activities of Daily Living Scale (BADLS, on which scores range from 0 to 60, with higher scores indicating greater impairment) compared with baseline [286]. In most clinical studies of donepezil, patients treated with donepezil maintained or improved cognition and overall dementia severity in contrast to placebo-treated patients who showed a decline in cognition [287].
The accumulation of Aβ in the brain is part of the pathology of AD, and in animal studies, donepezil reduces brain Aβ accumulation [288, 289]. However, using 11C-Pittsburgh compound B, a clinical study of AD patients receiving donepezil (5 or 10 mg) for at least 1 year showed that there was no reduction in Aβ accumulation at either dose [290]. Another symptom of AD, other than memory loss, is agitation [245]. Despite appearing to be effective in the cognition portions of clinical trials, donepezil does not appear to be effective at managing agitation in AD patients [291].
2.8 Eliprodil
Eliprodil (Fig. 2) is an NMDA antagonist that selectively inhibits the NR2B (GLUN2B) and is a σ1R agonist. Eliprodil failed a phase III clinical trial to treat acute ischaemic stroke [292], and has also been trialled for schizophrenia [293]. Eliprodil has a moderate affinity for the σ1R (Ki= 630 nM) [294].
2.8.1 Parkinson’s Disease
In a pre-clinical study of eliprodil, a low dose of eliprodil stimulated locomotion in mouse PD models, whereas a high dose had more sedative action. In the same study of reserpine-treated mice, eliprodil reversed akinesia, although this was not statistically significant. Furthermore, eliprodil failed to improve motor recovery from the DA D1 agonist SKF38393, or the DA D2 agonist RU24213, and inhibited the motor stimulation induced by L-DOPA [295]. Eliprodil has been clinically trialled for movement disorders, including PD (NCT00001929), although the data were never published. Based on this limited evidence, eliprodil is unlikely to be an effective PD treatment.
2.8.2 Multiple Sclerosis
Eliprodil has been reported to exhibit a neuroprotectant effect against toxic insults. A study by Demerens et al. [296] has indicated the ability of eliprodil to induce myelination in cellular models, suggesting that the molecule can be of therapeutic interest for the treatment of MS.
2.9 Fabomotizole
Fabomotizole (Afobazole) (Fig. 2) was launched in Russia in the early 2000s as an anxiolytic drug. As well as acting on the melatonin receptor 1 and 3 (MT1 and MT3), Afobazole has GABAergic [297], NGF and BDNF-releasing properties [298]. Afobazole is a mixed σ1/2R ligand [299].
2.9.1 Alzheimer’s Disease
Afobazole has been shown to inhibit Aβ25-35-induced calcium increase and nitric oxide production in both rat cortical neurons and microglial cultures [300, 301]. Afobazole shows some promise as a σ1/2R ligand; however, its use in AD is still in the pre-clinical stage, and there appear to be no current or planned clinical trials.
2.10 Fenfluramine
Fenfluramine (Fintepla) (Fig. 2) is a positive modulator at the σ1R [89], with an affinity of 266 nM (Ki guinea pig brain) [302]. It also acts to cause the release of serotonin by disrupting the vesicular storage of the neurotransmitter and reversing serotonin transporter function.
2.10.1 Developmental and Epileptic Encephalopathies
The serotonin activity of fenfluramine fails to explain its efficacy in Dravet syndrome (a genetic epilepsy characterized by temperature-sensitive/febrile seizures). Furthermore, fenfluramine could potentiate the BiP dissociating effects of σ1R agonist PRE084 (although it did not affect BiP dissociation by itself), indicating that fenfluramine is a positive modulator of the σ1R rather than a σ1R agonist. However, while the positive modulator SOMCL-668 reduced epileptic activity in zebrafish, the σ1R agonist PRE084 inhibited fenfluramine-mediated anti-epileptic activity in zebrafish models [303]. This may be explained by the biphasic nature of many σ1R ligands in vivo [304].
In a clinical trial of fenfluramine (NCT03355209) in patients with Lennox–Gastaut syndrome (another developmental and epileptic encephalopathy), fenfluramine, compared with placebo, decreased drop seizures and may be particularly good for patients who experience generalized tonic–clonic seizures [304]. In another pair of fenfluramine studies in Dravet syndrome, where the two identical studies were merged (NCT02682927 and NCT02826863), fenfluramine provided a significantly greater reduction in convulsive seizure frequency compared with placebo [305]. In total, 119 patients, with a minimum number of convulsive seizures per 4-week period for 12 weeks before screening, were randomized to receive placebo, fenfluramine 0.2 mg/kg/day or fenfluramine 0.7 mg/kg/day. The study met its primary efficacy endpoint, with 0.7 mg/kg/day showing a 62% greater reduction in mean monthly convulsive seizure frequency than placebo. Furthermore, the low dosage of fenfluramine (0.2 mg/kg/day) showed a 32% reduction in mean MCSF compared with placebo [305].
Another study of fenfluramine provided a clinically meaningful reduction in convulsive seizure frequency in most patients in a real-world study (71.1% of patients had a greater than 50% reduction of convulsive seizures, 53.3% had a more than 75% reduction and 11.1% were seizure free) [306]. Several other studies are ongoing, and studies with data yet to be published are detailed in Table 3.
| Study title | Conditions | Primary outcome measures | Study results | Phase | Trial number |
|---|---|---|---|---|---|
| Fenfluramine for adult Dravet patients | Dravet syndromeDravet syndrome, intractable | Seizure reductionComparing the proportion of participants with >50% reduction in the monthly countable convulsive seizure frequency between 4 week baseline period (observational phase) and after 12 week treatment phase using seizure diary | No results posted (last update 2022) | 3 | NCT05560282 |
| Treatment of Dravet syndrome with fenfluramine (expanded access protocol) | Dravet syndrome | Evaluate the safety of fenfluramine | No results posted (last update 2020) | Unknown | NCT04437004 |
| A study to investigate the long-term safety of ZX008 (fenfluramine hydrochloride) oral solution in children and adults with epileptic encephalopathy including Dravet syndrome and Lennox–Gastaut syndrome | Dravet syndromeLennox–Gastaut syndromeEpileptic encephalopathy | Evaluate the safety of fenfluramineChange in convulsive seizure responseChange in cognitive CGI by parent/caregiver and investigatorChange in behavioral CGI by parent/caregiver and investigatorChange in motor CGI by parent/caregiver and investigatorChange in clinical global impression by parent/caregiver and investigator | No results posted (last update 2023) | 3 | NCT03936777 |
| Fenfluramine for the treatment of different types of developmental and epileptic encephalopathies: a pilot trial exploring epileptic and non-epileptic outcomes (FENDEEP) | Refractory epilepsySYNGAP1 encephalopathySTXBP1 encephalopathy with epilepsyInv Dup(15) EncephalopathyMultifocal or bilateral malformations of cortical developmentContinuous spike and waves during slow sleep | Seizure frequency, measured using a seizure diary | No results posted (last update 2022) | 4 | NCT05232630 |
Ongoing clinical trials of fenfluramine
CGI clinical global impression
2.11 Fluoxetine
Fluoxetine (Fig. 2) is an SSRI, broadly administered as an antidepressant. Besides its use as an antidepressant, fluoxetine has been suggested for many off-label uses, such as cancer treatment [307] and COVID-19 [308]. Fluoxetine can regulate the inflammatory response by reducing tumour necrosis factor (TNF)-α; interleukin (IL)-1β, -2 and -6; and interferon (INF)-γ, while increasing IL-2, -4 and -10 [309]. Fluoxetine has an affinity for the σ1R of 214–240 nM [310].
2.11.1 Multiple Sclerosis
In a pilot study with relapsing MS patients, fluoxetine reduced the formation of new-enhancing inflammatory lesions in the brain [311]. Fluoxetine attenuated microglial activation and protected oligodendrocytes from apoptosis, preventing myelin loss through the inhibition of c-Jun [312]. Oral administration of fluoxetine (NCT01910259) in MS patients was found to reduce axonal degeneration as measured by proton magnetic resonance spectroscopy [309]. However, in secondary progressive MS individuals, fluoxetine treatment did not exhibit any significant changes in brain atrophy [313–317].
2.12 Fluvoxamine
The SSRI fluvoxamine is a strong σ1R agonist and has been proven to have strong efficacy against depression. Recent studies indicate it may help treat COVID-19 [318]. It could potentiate neurite outgrowth in PC12 cells in a σ1R-dependent mechanism, indicating its neuroprotective potential [319]. In an open-label study, the administration of fluvoxamine exhibited antidepressant activity in MS individuals [320].
2.12.1 Multiple Sclerosis
Fluvoxamine increased the proliferation of neural stem cells and facilitated their differentiation into oligodendrocytes through the Notch signalling pathway. Further, in an experimental autoimmune encephalitis animal model, fluvoxamine treatment reduced the severity of the condition by inhibiting IFN-γ, and reducing inflammatory infiltration and demyelination, suggesting therapeutic potential against MS [321].
2.13 Ketamine
Ketamine (Fig. 2) is an NMDA receptor antagonist acting to block the open ion channel similarly to memantine. Ketamine also has a moderate affinity for the σ1R (Ki = 139.60 μM). It can potentiate PC12 cell neurite outgrowth in response to NGF (a common assay of σ1R agonism) in a bell-shaped dose-response curve (also a common feature among σ1R agonists), with the optimal concentration being 1 nM [322]. Furthermore, NE100 (a potent σ1R antagonist) could inhibit the neurite outgrowth potentiation. As we have discussed above, developing new neuron connections in the hippocampus is important in the progression and treatment of AD. Treatment of mice with low-dose ketamine (8 mg/kg) reduced behavioural abnormalities and improved cognitive function in mouse models of Rett syndrome (a neurodegenerative disease discussed further below). Furthermore, 10 mg/kg ketamine improved cognitive flexibility in rats subjected to repeated restraint stress [323].
2.13.1 Alzheimer’s Disease
Ketamine has been shown to prevent lipopolysaccharide (LPS)-induced TNF-α production in glial cells, astrocyte cultures and microglial cultures [324], suggesting that ketamine can prevent neuroinflammation.
A clinical trial (NCT02433041) investigating ketamine to prevent post-operative delirium found, as part of the secondary outcomes, a significant decrease in inflammatory markers with the use of ketamine post-operatively when compared with the placebo [325]. This suggests ketamine has neuroprotective action on neurons, glial cells and astrocytes.
Several studies have shown that low doses of ketamine helped improve visual memory, simple working memory and complex working memory in patients with treatment-resistant depression [326, 327]. Depression is a common symptom of AD; furthermore, depression is linked to AD development [328]. Ketamine relieves depressive symptoms within hours of treatment, especially in cases where other antidepressants have failed [329]. In a case report of a 76-year-old female AD patient suffering from depression who had not responded to conventional antidepressant therapy, the patient was reported to have recovered from severe treatment-resistant depressive decline after only two doses of ketamine [330].
Recently, a meta-analysis review of 25 randomized controlled trials that included 14 commonly prescribed antidepressants failed to show efficacy in AD patients [331]. Thus, with ketamine’s potential for neuroprotection and its efficacy in treating depression, it is possible that ketamine could be a useful intervention in treating depression in AD. However, studies of prolonged treatment (3 months) with ketamine in mice and monkeys show an increase in tau hyperphosphorylation, a key part of AD pathology, before the formation of neurofibril tangles (NFTs). The study did not show Aβ plaque formation, another pathological hallmark of AD [332].
There are several safety issues concerning ketamine use over long-term treatment (discussed below regarding PD, although many concerns also apply to AD). Therefore, further study of the efficacy and safety of ketamine at various doses and over short- and long-term treatment is required before ketamine can be seriously considered as a treatment for AD.
2.13.2 Parkinson’s Disease
Ketamine in combination with xylazine (α2 adrenergic agonist) has been studied pre-clinically in rat models of PD (MPTP and 6-OHDA). The ketamine/xylazine (85/3 mg/kg) combination given before the MPTP or 6-OHDA significantly reduced the loss of tyrosine hydroxylase immune-stained cells. Furthermore, ketamine/xylazine treatment reversed the amnestic effects of the two neurotoxins, with ketamine/xylazine-treated rats performing the same as control rats in the hidden platform test (MPTP- or 6-OHDA-treated rats showed a 10-second latency in finding the hidden platform) [333]. Another study of rats subjected to 6-OHDA showed reduced mobility time in the forced swim test (suggesting depressive-like behaviour). Rats treated with ketamine for 28 days (starting 21 days after the brain lesions were created with 6-OHDA) increased mobility times in the water at all doses of ketamine. Furthermore, ketamine also reduced symptoms of anhedonia (caused by the 6-OHDA lesions) measured using the sucrose preference test. The same study also investigated short-term memory in the rats using the social recognition test, and ketamine significantly (at all doses 5–15 mg/kg) reversed the amnesic effects of 6-OHDA treatment [334]. In a mouse model of PD using MPTP to induce PD-like symptoms in the mice, ketamine (8 mg/kg) improved the coordinating movement, learning and memory ability of PD model mice.
Furthermore, ketamine also prevented tyrosine hydroxylase-positive neurons from being attacked by MPTP, therefore, protecting nigra dopaminergic neurons. MPTP treatment of mice significantly increased mTOR expression and lowered the expression of several autophagy markers (LC3-II, Beclin1, Parkin and PINK). Ketamine restored expression levels of LC3-II, Beclin1, Parkin and PINK1 to levels of the untreated controls, and reduced the expression of mTOR. This indicates that the subanaesthetic dose of ketamine can restore autophagy in the PD mouse model [335].
In a case report of an 88-year-old female patient with PD exhibiting severe bilateral dyskinesia who required surgery, ketamine [20 mg intravenous (IV)] followed a dose of midazolam (1 mg IV) (to prevent the possible psychotomimetic effects of ketamine). It resolved her severe tremor both at rest and during intentional movement. Following the ketamine, the patient remained responsive, communicative and able to follow commands. Several days later, during post-operative care, she remembered receiving the pre-operative ketamine with no adverse consequences [336]. Two previous cases have used ketamine to permit surgery in cases of PD, both of which were completed with this drug combination resulting in no major side effects [337]. Ketamine has now entered phase 2 clinical trials for treating depression in PD (NCT04944017) and treating levodopa-induced dyskinesia (NCT04912115). Both studies are still ongoing.
There are several possible drawbacks to the use of ketamine in the treatment of depression and levodopa-induced dyskinesia in PD. In treating depression (in general), ketamine IV injection relieves depression symptoms for 1–2 weeks, with some patients reporting only a few days. One study of eight patients where ketamine (0.5 mg/kg IV) was administered up to every two weeks showed promising results [338]. Furthermore, ketamine dosing requires injections, as it has poor bioavailability and a high first-pass hepatic metabolism, thus requiring hospital/clinical visits. Trips to the hospital/clinic every 3–7 days for an IV infusion of ketamine are not practical. Ketamine also has psychotomimetic effects and dissociative symptoms (although these tend to dissipate within 4 hours of treatment) [339] and has the potential for addiction [340]. There are also concerns about the long-term safety of ketamine treatment as transient tachycardia, hypertension, neurotoxicity, bladder toxicity and tolerance are reported with repeated ketamine infusion [341].
2.13.3 Multiple Sclerosis
In the experimental autoimmune encephalitis mice model, (R)-ketamine reduced microglial activation, preserved blood–brain barrier (BBB) integrity and ameliorated the demyelination [342]. Further, under cuprizone-induced demyelination in mice, (R)-ketamine facilitated remyelination through activating TrkB and altered the abnormal amount of Eisenbergiella massiliensis in the gut microbiota, which is positively correlated with demyelination [343]. Oral administration of ketamine in a 60-year-old woman with MS reduced neuropathic pain (allodynia) associated with the disease [344]. In a patient with treatment-resistant depression associated with MS, IV ketamine administration significantly reduced the depression index score with no worsening of her condition over a two-year observation period [345]. Likewise, ketamine also improved fatigue, which is associated with MS, in an active placebo-controlled double-blind trial (NCT03500289), suggesting its beneficial effects. Many of the drawbacks mentioned above regarding ketamine and PD also apply here.
2.13.4 Rett Syndrome
In a pre-clinical study, daily treatment with ketamine reduced Rett syndrome-like symptoms and extended the life span of MeCP2-null mice without adverse side effects. Furthermore, cortical processing and connectivity were fully restored to wild-type levels [346]. Functional deficits in forebrain circuits in mutant MeCP2 mice can be restored by ketamine treatment [347].
Two clinical trials (phase 1 and phase 2) are registered at www.clinicaltrials.gov(NCT02562820 and NCT03633058). The phase 1 safety study was terminated due to funding withdrawal, and the phase 2 study is listed as complete; however, there are no listed results or peer-reviewed publications yet. As well as the drawbacks of ketamine described above, ketamine may also provoke epileptic discharges in some patients with a seizure disorder [348].
2.14 Memantine
Memantine (Ebixa, Axura, Namenda, Akatinol) (Fig. 2) is a low–moderate affinity NMDA receptor antagonist often prescribed for AD patients who do not tolerate AChEI drugs such as donepezil [349]. Memantine, in combination with donepezil, is approved as a treatment for AD in many countries [350]. Memantine is prescribed for moderate-to-severe AD. It acts as an uncompetitive channel blocker of the NMDA receptor, i.e. memantine does not interact with the binding sites of the NMDA agonists, rather it fits into the channel, competing with magnesium ions, blocking ion flow through the channel into and out of the cell. It is somewhat surprising that an antagonist of the glutamate receptor can benefit AD symptoms since AD sees a reduction in glutamate and glutamate receptors [351].
2.14.1 Alzheimer’s Disease
It has been hypothesized that several mechanisms mediate the beneficial effects of memantine in AD, including decreasing synaptic ‘noise’ from excessive NMDAR activation and reducing the excitotoxicity [351, 352] or inhibiting Aβ production or toxicity [353]. It is hypothesized that memantine’s fast on/off pharmacokinetics allow for the physiological activation of NMDARs, while at the same time inhibiting pathological overactivation because of its mechanism of NMDAR inhibition.
The σ1R has been shown to play a role in regulating the NMDA receptor and has been well documented [41, 354, 355]. For example, σ1R KO impairs neurogenesis in the hippocampus of adult mice via the downregulation of the NMDA receptor [356]. Furthermore, activation of the σ1R results in the upregulation and transport to the plasma membrane of NMDA receptors [357].
Memantine, like other NMDA ion-channel blockers (amantadine [5] and ketamine [358], among others), has some affinity for the σ1R. However, the affinity of memantine is rather low (2.5 µM) [200], and thus the σ1R is unlikely to play a full role in memantine’s therapeutic effects. However, since activating the σ1R receptor modulates NMDA function and upregulates NMDA expression and trafficking to the plasma membrane, it is possible that the combination of drugs such as memantine and a σ1R agonist could be beneficial in AD. Monotherapy with memantine has been clinically trialled in multiple studies and is generally safe and well tolerated, plus it has beneficial effects compared with a placebo. A meta-analysis of six placebo-controlled phase 3 trials supports the clinically relevant efficacy of memantine in patients with moderate-to-severe AD [359]. The combination therapy of memantine and donepezil has been trialled in several studies with mixed results (NCT00866060, NCT02580305, NCT03316898 and NCT02162251). Multiple studies have identified significant improvements in preventing cognitive decline when memantine is added to stabilize the treatment of patients with stable donepezil treatment for moderate-to-severe AD [360,361,362,363,364]. However, other studies have been less convincing, with the data suggesting only a mild to no benefit to the combination therapy over monotherapy with either donepezil or memantine [365].
2.14.2 Parkinson’s Disease
Memantine is not used in treating the motor symptoms of PD. However, it has been trialled for treating PD-related dementia [366, 369]. Memantine is relatively safe and well tolerated in PD-related dementia treatment, and there appear to be some studies with positive outcomes [367].
However, a meta-analysis study of ten trials indicates that memantine does not improve scores on the MMSE, whereas cholinesterase inhibitors did [370]. The study did find that there was a reported improvement in global impression but not cognitive function. The clinical trials are listed in Table 4.
| Study title | Primary outcome measures | Study results | Phase | Trial Number |
|---|---|---|---|---|
| Namenda (memantine) for non-motor symptoms in Parkinson’s disease | UPDRSAnalyses will be computed for the categorical DV: Global Tremor Assessment by examiner | Memantine was well tolerated in Parkinson’s disease with few adverse events. However, specific measures of sleepiness, fatigue, depression and attention did not significantly improve [367] | 4 | NCT00646204 |
| Parkinson’s disease evaluated by PET and the effect of memantine | Decreases in cerebral blood flow and oxygen metabolism in these areas will be the consequence and this can be detected by PET | No results posted (last update 2006) | N/A | NCT00375778 |
| Study of memantine for gait disorders and attention deficit In Parkinson’s disease | Stride length by gait analysis with an optoelectronic system | In the memantine treated group, statistically significantly better results (versus placebo) were observed for the overall UPDRS score [368] | 4 | NCT01108029 |
| Memantine versus placebo in Parkinson’s disease dementia or DLB | Assess the global benefits of memantine compared with placebo in outpatients with a diagnosis of PDD or DLB (mild to moderate severity) over a 6 month period | Patients with DLB treated with memantine showed greater improvement according to Alzheimer’s disease cooperative study (ADCS)-clinical global impression of change scores than those who received placebo. No significant differences were noted between the two treatments in patients with PDD [366] | 4 | NCT00855686 |
| Memantine for treatment of cognitive impairment in patients with Parkinson’s disease and dementia | DRS Memory SubscoreCIBIC–Plus Score | DRS scores appeared to improve with a score of 1.6 (95% CI −0.13 to 3.33) in memantine treated compared with −1.4 (95% CI −3.19 to 0.39) in placebo treated. There was also an apparent improvement in the CIBIC–Plus Score in the memantine treated groupe | N/A | NCT00294554 |
| Efficacy and safety of memantine for PDD and dementia with Lewy bodies (DLB) (MEMPDD) | Clinical Global Impression of Change | No results posted (last update 2015) | 2 | NCT00630500 |
| Monitoring anti-dementia drugs by serum levels (MONANTI) | Change of MMSEChange of ACEChange of NPI-QChange of DADChange of clinical serum level of memantineChange in GDS | No results posted (last update 2020) | 4 | NCT04117178 |
Clinical trials of memantine for Parkinson’s disease and dementia-related symptoms
ACE Adenbrooke’s Cognitive Exam, DV dependent variable, DAD Disability Assessment for Dementia, DRS Dementia Rating Scale, DLB dementia with Lewy bodies, GDS Geriatric Depression Scale, MMSE Mini Mental State Examination, NPI-Q Neuropsychiatric Inventory Questionnaire, PDD Parkinson’s disease dementia, PET positron emission tomography, UPDRS Unified Parkinson Disease Rating Scale
2.15 Naluzotan
Naluzotan (Fig. 2) acts as a selective and potent 5-HT1A receptor partial agonist, and it has been claimed that it binds to and activates the σ1R, although the evidence for this is weak. Naluzotan has a claimed affinity of 100 nM for the σ1R; however, few references to the σ1R of naluzotan can be found, with no binding data to back up the claims [371]. It has been trialled for depression and anxiety, with the study suggesting that naluzotan is relatively safe and higher doses of naluzotan may be effective in anxiety and depression [372].
2.15.1 Developmental and Epileptic Encephalopathies
Naluzotan has been tested for its effects in localization-related epilepsy (NCT01281956) [373], with the data suggesting that there is no significant difference between placebo and naluzotan; furthermore, the number of seizures in the naluzotan group appeared to be higher than that of the placebo and the trial was terminated.
Given the lack of evidence for the role of the σ1R in the pharmacology of naluzotan, and that it had no effect of seizure frequency when trialled, naluzotan is unlikely to benefit DEE patients.
2.16 Phenytoin
Phenytoin (Dilantin, amongst others) (Fig. 2) has long been used clinically for seizures [374]. The primary mechanism for phenytoin is thought to be the inhibition of voltage-gated sodium channels [375]. Phenytoin is a non-selective allosteric modulator of the σ1R. There is much evidence that the σ1R interacts with and modulates the function of various ion channels, including calcium channels, with many studies indicating that the σ1R acts to negatively regulate calcium movement [376,377,378,379].
Phenytoin is a positive modulator of the σ1R as it increases σ1R agonist binding to the receptor when co-administered with a σ1R agonist. Furthermore, it can be used to distinguish between σ1R agonists and antagonists as phenytoin does not increase antagonist binding [380].
2.16.1 Developmental and Epileptic Encephalopathies
Many clinical studies have investigated phenytoin (a total of 123 listed on www.clinicaltrials.gov, of which 36 involve seizures). However, phenytoin is considered a last resort for treating encephalopathy since the severe side effects of phenytoin maintenance therapy, especially cognitive impairment, are undesirable in these already cognitively impaired patients [381]. There has been one case report that suggests phenytoin treatment in a two-year-old male with epileptic encephalopathy that did not improve with phenobarbital, pyridoxine, pyridoxal phosphate and levetiracetam showed dramatic response to an IV loading dose of phenytoin, followed by a transfer over to carbamazepine to prevent seizure relapses because of the difficulty in maintaining appropriate blood levels of phenytoin [382].
2.17 PRE-084
The selective σ1R agonist, 2-(4-morpholino ethyl)1 phenylcyclohexanecarboxylate (PRE-084) (Fig. 2) contains the three pharmacophoric moieties for a σ1R ligand: a hydrophobic cluster, an amine group and an intermediate chain [383], and has an affinity of 2.2 nM. PRE-084 has been a very useful pharmacological tool in the discovery of other σ1R ligands [384]. Although thought to be specific to the σ1R, a study has shown that it can have some off-target effects. PRE-084 could elicit an enhanced barrier function of human umbilical vein endothelial cell (HUVEC) monolayers in a dose-dependent fashion, and even with significant siRNA knockdown of the σ1R, PRE-084 maintained the barrier function effect, meaning that either only a low occupancy of σ1R is required for the barrier function effect, or that another receptor is involved [385].
2.17.1 Alzheimer’s Disease
PRE-084 has many characteristics that could make it a potential treatment for AD. Pre-clinical data have shown that PRE-084 can attenuate the blood–brain barrier (BBB) disruption caused by Aβ. The study showed that PRE-084 protected the integrity of the BBB by preventing the deposition of Aβ (insoluble) Aβ1–40 and Aβ1–42 levels in the brains of mice, and increasing vascular endothelial growth factor (VEGF) and ApoE receptor [386]. PRE-084 also increases the expression of glial cell line-derived neurotrophic factor (GDNF) [387] and BDNF [388].
The hippocampal region of the brain is responsible for learning and memory and is one of the first regions to show damage in AD [389]. Neurogenesis occurs in the adult hippocampus, although with age, this begins to slow [390]. A decreased rate of neurogenesis, increased destruction of mature neurons, and increased inflammation could contribute to the cognitive symptoms seen in AD. It is thought that this damage in the hippocampus can occur up to 10 years before cognitive symptoms appear in the patient. Studies using Aβ1–42 [391] or Aβ25–35[392] injections into mouse brains have shown reduced proliferation and neurite growth, increased death of newly formed cells and an increase in the number of immature cells in mice brains. PRE-084 treatment promotes neurogenesis in mice treated with Aβ1–42 or Aβ25–35[391, 392]. Furthermore, the density of hyper-reactive astrocytes was reduced by the administration of PRE-084 [391], suggesting PRE-084 may play an anti-inflammatory role. PRE-084 could also increase mushroom dendritic spine formation in the hippocampus of transgenic rats expressing presenilin-1 (PS1) and human APP(Swe) [393].
There has been no clinical study registered or completed with PRE-084 for AD or any other neurodegenerative disease, and there are no data on the safety or pharmacokinetic profile for PRE-084 in human trials. PRE-084 is a well-known selective σ1R agonist [380] and has been used over the past 30 years as an important prototypical drug to uncover potential therapeutic benefits of the σ1R. However, it has yet to be investigated clinically, possibly due to its potential off-target effects, which are thought to increase with dose [384].
2.17.2 Amyotrophic Lateral Sclerosis/Motor Neuron Disease
PRE-084 protected spinal cord organotypic culture explants from glutamate toxicity and induced the elongation of neurites in motor neurons through the activation of PKC [394]. Administration of PRE-084 in adult rats prevented the loss of axotomized motoneurons upon spinal root avulsion injury with decreased astroglial immunoreactivity, alongside increased BiP and GDNF expression in motor neurons and astrocytes, respectively [387]. In SOD1G93Amice, PRE-084 treatment improved locomotor performance, preserved motor neurons, extended survival of animals and modulated NMDA receptor calcium influx through the PKC pathway [395,396,397]. In wobbler mice, chronic treatment of PRE-084 increased BDNF expression, reduced reactive astrocytes and induced the survival of motor neurons indicating σ1R and their agonists can also be used for treating non-SOD1-linked ALS [398]. Nrf-2 co-localization with σ1R has been reported in photoreceptor cells, implying Nrf-2 could be directly targeted by σ1R [399]. However, possibly due to a difference in the study design, there are also data suggesting that NRF-2 does not interact with the Nrf-2 [400]. PRE-084 treatment rescued motor neuron defects in TDP43G348C zebrafish by enhancing ATP generation, boosting the unfolded protein response (UPR) and activating the Nrf-2 pathway [401].
2.17.3 Huntington’s Disease
PRE-084 has been tested pre-clinically as a therapeutic agent for HD. A study in 2013 [402] showed that PRE-084 promotes cell survival and counteracts the detrimental effects of mutant N-terminal huntingtin protein in neuronal PC6.3 cells. PRE-084 increased cellular antioxidants through the NF-κB pathway (a pathway normally compromised by the mutant huntingtin proteins), supporting the future use of σ1R agonists as therapeutics for HD.
Treatment of transgenic cortical cultures from mice expressing 128 CAG repeat expansion in the Htt gene (YAC128 transgenic mice) with PRE-084 (100 nM) restored synaptic connections between the cortical and striatal neurons but did not affect their number in the cultures when compared with wild-type mice; furthermore, this was reversed with the σ1R antagonist NE-100 [403].
2.17.4 Wolfram Syndrome
PRE-084 has been shown in a pre-clinical study to attenuate learning deficits in male Wfs1ΔExon8 mice (a transgenic WS model) and reduce anxiety in female Wfs1ΔExon8 mice [197]. In the same study, wfs1abKO (zebrafish larvae with the Wfs1 gene knocked out) showed a clear locomotor alteration when their response to light variation was tested. PRE-084 treatment (0.1–10 µM) in the fish water resulted in a dose-dependent decrease in the hyperlocomotion response, back down to the level of the wild-type larvae (Wfs1abWT) [197]. Furthermore, these effects were reversed by the σ1R antagonist NE-100, suggesting that the σ1R is responsible for these beneficial effects.
2.18 Pridopidine
Pridopidine (Fig. 2), developed by Prilenia Therapeutics, is a drug currently in the late stages of clinical trials for PD, ALS and HD [52]. Pridopidine is a selective σ1R ligand with low affinity for DA D2/D3 receptors, the adrenergic a2C receptor and the σ2R [404], and has the potential as an AD therapy. At clinically relevant doses, positron emission tomography (PET) imaging studies in rats and humans show high occupancy of the σ1R, with no or negligible occupancy of the D2/D3 receptors [405, 406].
2.18.1 Alzheimer’s Disease
AD cognitive impairment results from damage to synaptic connections and neuronal atrophy. The plasticity of dendritic spine structures in developing neurons underlies memory formation, as thin spines convert into mushroom spines during information storage in response to long-term potentiation (LTPs) [407,408,409]; mushroom spine deficiency may reflect learning and memory defects in models of AD. Pre-clinical studies have shown that pridopidine stabilizes mushroom spines in mouse models of AD. In PS1 knock-in (PS1-KI) mouse hippocampal cultures and wild-type cultures treated with Aβ1–42, the percentage of mushroom spines was significantly reduced. This reduction could be prevented by 100 nM pridopidine. Furthermore, this was repeated in vivo in PS1-KI mice [63].
Pridopidine protects against NMDA-induced oxidative stress in primary hippocampal cultures and can increase mushroom dendritic spine formation in the hippocampus of transgenic rats expressing presenilin-1 (PS1) and human APP(Swe), as well as reduce deficits in spatial learning [393]. The study also showed that pridopidine induced ERK and AKT phosphorylation. Thus, the involvement of the mitogen-activated protein kinase/extracellular signal-regulated kinases (MAPK/ERK) and phosphoinositide 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/Akt/mTOR) signalling pathways, through which the σ1R may control the neuroprotective properties and synaptogenesis [393].
Pridopidine has yet to be studied clinically for AD; however, given the success of pridopidine in treating other neurodegenerative diseases (discussed further below) and its pre-clinical data in AD, it is possible that pridopidine could show some benefit in patients with AD.
2.18.2 Amyotrophic Lateral Sclerosis
Pridopidine targets σ1R in SOD1G93A ALS models (both in vitro and in vivo) and ameliorates the pathology by maintaining neuromuscular junction integrity and enhancing axonal transport through the activation of ERK pathway [410]. Also, cachexia and motor neuron deficits were prevented in SOD1G93A mice upon pridopidine administration [411]. One of the important steps during autophagy is the transport of transcription factor EB (TFEB) into the nucleus through POM121/KPNB1/importinβ1. The occurrence of hexanucleotide RNA expansion (HRE) because of defective C9orf72 gene interferes with the σ1R and POM121 interaction, causing reduced TFEB transport and autophagy. In NSC34-expressing (G4C2)31 RNA repeats potentiated σ1R chaperone activity, and enhanced POM121 and autophagy [412]. Under the HEALEY ALS Platform Trial umbrella, two ongoing phase 2/3 clinical trials are active for ALS patients to study pridopidine (as well as other drugs, such as zilucoplan, verdiperstat, CNM-Au8, SLS-005 Trehalose and ABBV-CLS-7262) on disease progression, respiratory function, muscle strength, bulbar function and survival (NCT04615923 and NCT04297683).
2.18.3 Huntington’s Disease
Treatment of YAC128 HD mice with pridopidine reduces anxiety and depression-like symptoms [413], prevents striatal medium spiny neuron (MSN) loss and facilitates Ca2+homeostasis and signalling at the ER via activation of the σ1R [97]. In both animal and human studies of HD, the BDNF pathway has been shown to be affected [414]. Under non-disease conditions, the Htt protein is involved in the transport of BDNF. However, the mutated version of Htt protein reduces BDNF release [415]. Furthermore, BDNF overexpression can rescue HD phenotypes in YAC128 mice [416]. This is particularly relevant to the σ1R, as σ1R activation has been shown in multiple studies to induce BDNF expression and activity [188, 416,417,418].
Pridopidine has undergone randomized, double-blind clinical trials for HD treatment [419, 420]. Pridopidine treatment was ineffective at 20 mg/day and 45 mg/day. However, the higher 90 mg/day improved movement ability measured by the total motor scores (TMS) [419].
Higher doses of pridopidine or longer treatment periods could show further improvements. However, another larger study failed to replicate the improvements in TMS. However, some improvements were seen at 26 and 52 weeks in an exploratory assessment of rater-independent Q-Motor and total functional capacity (TFC) using a dose of 45 mg BID (90 mg/day) [421].
Furthermore, an open label study of pridopidine covering 60 months showed that 45 mg BID (90 mg/day) can maintain stable TFC and TMS, when using historical placebo controls as a comparison [422]. Pridopidine, although not a cure-all drug, does show some promising improvements in some aspects of HD and is tolerable orally at doses up to 112.5 mg BID (225 mg/day), therefore, further investigation is warranted into its benefits for HD treatment.
2.18.4 Parkinson’s Disease
Pridopidine has been investigated pre-clinically for its use in PD. In a mouse model of PD where 6-OHDA was used to create lesions that resulted in several PD-like symptoms in the mice, pridopidine significantly improved deficits in forelimb use, offered significant protection of nigral DA cell bodies and increased dopaminergic fibre density in the striatum. In σ1R KO mice, pridopidine did not protect against 6-OHDA-induced lesions. The study also showed the upregulation of GDNF, BDNF and phosphorylated ERK1/2 in the striatal region of the brain [226]. In a similar rat model, pridopidine decreased L-DOPA-induced sensitized locomotor activity while permitting a beneficial motor-stimulant effect of the L-DOPA [423]. In primates [macaques treated with the neurotoxin 1-methyl-4-phenyl-l,2,3,6-tetrahydropyridine (MPTP)], Pridopidine produced a dose-dependent reduction in dyskinesia caused by L-DOPA without modulating the anti-parkinsonian benefit of L-DOPA [302].
Dyskinesia is involuntary hyperkinetic movements, and the risk of developing dyskinesia is approximately 25–40% after 4–6 years of levodopa therapy [424]. It is thought that L-DOPA does not cause the development of dyskinesia in PD. Rather it sensitizes the nigrostriatal system, since multiple clinical studies have shown that delaying L-DOPA treatment does not prolong the time before dyskinesia onset [425]. In a clinical trial of pridopidine in PD patients with dyskinesia (NCT03922711), pridopidine appeared to reduce Unified Dyskinesia Rating Scale (UDysRS) scores compared with the placebo after 5 weeks (the trial was terminated due to the COVID-19 outbreak). At time of writing, this is the only trial of pridopidine concerning PD, and, unfortunately, this trial was stopped early due to COVID, as the results looked promising.
2.19 Rivastigmine
Rivastigmine (Fig. 2) is a therapeutic agent that acts as an AChEI and is used in treating mild-to-moderate AD [426]. Rivastigmine is also thought to be active at the σ2R [29].
2.19.1 Alzheimer’s Disease
Rivastigmine enhances NGF-induced neurite outgrowth in PC12 cells via σ1R and σ2R since antagonists of each were both required to block the NGF-induced neurite outgrowth fully. Furthermore, σ1R and σ2R knockdown also abolished NGF-induced neurite outgrowth in PC12 cells [427].
Rivastigmine provides meaningful management of symptoms, maintains cognitive ability and daily function, and prevents some behavioural problems in AD [428,429,430].
2.20 SAS0132
SAS0132 (Fig. 2) is a novel potent σ2R ligand with a nine-fold selectivity over the σ1R. SAS0132 has an affinity of 90 nM for the σ2R, acting as a σ2R antagonist reversing intracellular calcium influx in human SK-N-SH neuroblastoma cells in response to σ2R agonist DKR-1051 [431].
2.20.1 Alzheimer’s Disease
In transgenic C. elegans overexpressing the APP gene, SAS0132 reduced the age-dependent neurodegeneration compared with controls. Furthermore, in the Thy-1 hAPPLond/Swe+transgenic mouse model of AD, SAS0132 improves cognitive performance [431]. SAS0132 is a promising σ2R antagonist with interesting results in pre-clinical studies; however, it has yet to enter clinical trials.
2.21 T-817 MA
T-817MA (edonerpic maleate) (Fig. 2) is a σ1R ligand with an affinity (Ki) of 16 nM. It has been shown to induce the outgrowth of neurites in cultured rat cortical neurons and protect against sodium nitroprusside-induced ROS production and cell death, which could be reversed by the σ-1 R antagonist BD1047 [432].
2.21.1 Alzheimer’s Disease
In animal studies of AD, edonerpic maleate ameliorated the deficits in adult neurogenesis and spatial memory in rats infused intra-cerebroventricularly (ICV) with Aβ peptide and showed neuroprotective effects on learning deficits induced by continuous infusion of Aβ peptide (1–40) [433, 434]. The doses required to see a therapeutic effect in these studies are higher than the reported Ki of 16 nM, with doses of 0.5–1 µM required to see positive effects. This possibly rules out the σ-1 R as the drug target of edonerpic maleate. However, this phenomenon is seen with other σ1R ligands, including IPAG and dipentylammonium [6, 156]. It is possibly explained by a non-sigmoidal dose-response curve, such as a bell-shaped or a multiple-binding sites curve [6].
In phase 1 clinical trials, edonerpic maleate was well tolerated at a single dose of up to 896 mg and once-daily doses of 672 mg for up to two weeks [435]. An initial phase 2 proof of concept study (NCT00663936) demonstrated the safety and tolerability of edonerpic maleate at a 224 mg/day dose for one year. Although it did not meet its primary and secondary outcome targets, post hoc analysis of a positive cognitive effect was seen in patients with moderate impairment (MMSE scores < 20) (reported in a poster at the Alzheimer’s Association international conference in 2013 [436]). When a larger phase 2 study of edonerpic maleate (NCT02079909) was conducted, it was shown to be generally safe and tolerable at the two doses studied (224 and 448 mg/day). However, in the primary measures of AD [AD Assessment Scale–Cognitive Subscale (ADAS–COG) and AD Cooperative Study–Clinical Global Impression of Change (ADCS–CGIC)], no significant difference was observed compared with the placebo. Furthermore, there were no significant improvements in secondary outcome, the AD Cooperative Study–Activities of Daily Living (ADCS–ADL) scale [435].
It is possible that since edonerpic maleate was tested at too high a dose, it fell on the far side of the bell-shaped dose-response curve, and therefore the positive effects were not seen. However, two doses were tested, one low and one high (224 and 448 mg/day), all of which put the expected serum concentrations well above that needed to occupy the σ1R. It is also possible that, like with blarcamesine, the heterogeneity of AD means that only a certain population of AD patients may benefit from edonerpic maleate treatment. However, further clinical study, including patient biomarker analysis, is required. Therefore, with the available (albeit limited) clinical evidence, it is likely that edonerpic maleate will not significantly help treat AD.
3 Conclusions
The σ1R interacts with multiple other protein complexes to modulate multiple signalling pathways and influence cellular health and survival (Fig. 3). This makes the σ1R an interesting target for the development of drugs for neurological diseases such as AD, HD, PD, MS and MND where neuron cell loss is a key part of the pathology. The fact that the expression of σ1R diminishes early on in AD and PD, and that there are genotypes for the σ1R that increase the risk of AD, PD and MS also add to the weight of evidence that the σ1R is important to these diseases and that σ1R ligands may be of benefit.
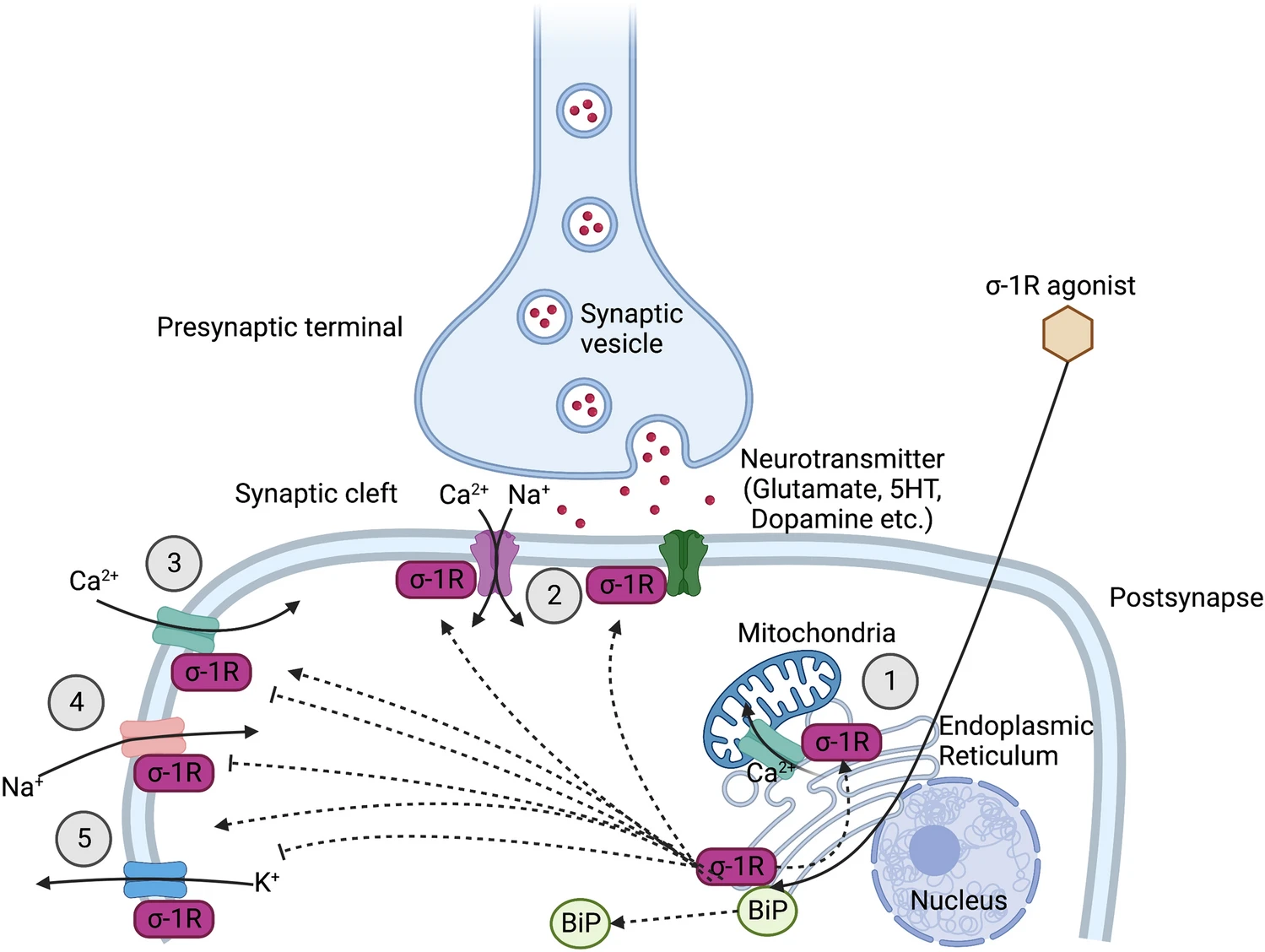
Some of the documented roles of the sigma-1 receptor in preventing neurological disease. (1) Activation of the sigma-1 receptor by an agonist result in the disassociation from binding immunoglobulin protein (BiP) and chaperoning of calcium channels at the mitochondria-associated ER membrane (MAM), resulting in calcium shuttling into the mitochondria and increased ATP production. (2) NMDA receptors are upregulated on sigma-1 activation and trafficked to the plasma membrane, and the sigma-1 receptor modulates NMDA receptor synaptic transmission and plasticity via SK channels. (3) Sigma-1 activation down-regulates calcium currents in L-type Ca2+ channels in retinal ganglion cells and upregulates calcium currents through L-type Ca2+ channels in the hippocampus. (4) Sigma-1 activation down-regulates Na+ currents through voltage-gated Na+ channels in intracardiac ganglion neurons. (5) Sigma-1 activation upregulates potassium currents through A-type K+ channels in neurohypophysial terminals and Xenopus oocytes. Parasympathetic intracardiac neurons CA1 field of hippocampus. Sigma-1 activation can upregulate human ether-a-go go K+ currents in Xenopus oocytes.
There have been vast strides taken in recent years for the early detection of diseases such as AD [437,438,439,440] and PD [441, 442], which means that new drug treatments targeting the σ1R may benefit more if they can prevent damage to the brain before the disease can progress. Drugs that target multiple targets, including the σ1R, such as blarcamesine or pridopidine in combination with Aβ antibodies, may prove to be effective in preventing neurological decline in AD. This possibly could result in the repurposing of older drugs, less specific to the σ1R in a multi-targeting and repurposing approach to drug development [52].
With the ever-evolving understanding of genetic variations and other biomarkers in the populations of people with these neurological diseases, there is a great opportunity to select medicines that are more likely to work in a specific population. For example, in AD, patients with the wild-type σ1R and higher scores on the MMSE fared better with blarcamesine than those with lower scores and a mutant σ1R.
The σ2R is often investigated concerning cancer treatment and imaging. However, in recent years, there has been a growing interest in both the σ2R and σ1R for treatments in neurological diseases. Despite their lack of homology, their similar binding profile and activity of the proteins mean both receptors need to be considered when designing novel drugs, since the σ1R agonists and σ2R antagonists are neuroprotective and σ1R antagonists and σ2R agonists are used in cancer study. In brief, with the current research and planned clinical studies, it is expected that there will be an advancement of new σ1R drugs for the treatment of the neurological disease including AD, HD and MS.