
Effects of sub-chronic, in vivo administration of sigma-1 receptor ligands on platelet and aortic arachidonate cascade in streptozotocin-induced diabetic rats
By Sándor Váczi, Lilla Barna, Krisztián Laczi, Ferenc Tömösi, Gábor Rákhely, Botond Penke, Lívia Fülöp, Ferenc Bogár, Tamás Janáky, Mária A. Deli, and Zsófia Mezei
Excerpt from the article published in Plos One on November 17, 2022, DOI: https://doi.org/10.1371/journal.pone.0265854
Editor’s Highlights
- The sigma-1 receptor (S1R), which modulates cellular functions, is expressed on both endothelial cells and platelets.
- Beneficial effects of S1R agonists have already been reported in sepsis, ischemic-reperfusion injury, stroke, and cardiovascular disease.
- S1R ligands may play a role in the regulation of cellular functions and local circulation by affecting AA metabolism at transcription, translation and enzyme induction levels.
- In diabetes mellitus, the cell-specific effects of S1R ligands have a compensatory role and aid in restoring physiological balance between the platelet and vessel.
Abstract
Background: Diabetes mellitus is a chronic metabolic disorder which induces endothelial dysfunction and platelet activation. Eicosanoids produced from arachidonic acid regulate cellular and vascular functions. Sigma-1 receptors (S1R) are expressed in platelets and endothelial cells and S1R expression is protective in diabetes.
Objectives: Our aim was to examine the influence of sub-chronic, in vivo administered S1R ligands PRE-084, (S)-L1 (a new compound) and NE-100 on the ex vivo arachidonic acid metabolism of platelets and aorta in streptozotocin-induced diabetic rats.
Methods: The serum level of the S1R ligands was detected by LC-MS/MS before the ex vivo analysis. Sigma-1 receptor and cyclooxygenase gene expression in platelets were determined by RT-qPCR. The eicosanoid synthesis was examined with a radiolabelled arachidonic acid substrate and ELISA.
Results: One month after the onset of STZ-induced diabetes, in vehicle-treated, diabetic rat platelet TxB2 and aortic 6-k-PGF1α production dropped. Sub-chronic in vivo treatment of STZ-induced diabetes in rats for one week with PRE-084 enhanced vasoconstrictor and platelet aggregator and reduced vasodilator and anti-aggregator cyclooxygenase product formation. (S)-L1 reduced the synthesis of vasodilator and anti-aggregator cyclooxygenase metabolites and promoted the recovery of physiological platelet function in diabetic rats. The S1R antagonist NE-100 produced no significant changes in platelet arachidonic acid metabolism. (S)-L1 decreased the synthesis of vasoconstrictor and platelet aggregator cyclooxygenase metabolites, whereas NE-100 increased the quantity of aortic vasodilator and anti-aggregator cyclooxygenase products and promoted the recovery of diabetic endothelial dysfunction in the aorta. The novel S1R ligand, (S)-L1 had similar effects on eicosanoid synthesis in platelets as the agonist PRE-084 and in aortas as the antagonist NE-100.
Conclusions: S1R ligands regulate cellular functions and local blood circulation by influencing arachidonic acid metabolism. In diabetes mellitus, the cell-specific effects of S1R ligands have a compensatory role and aid in restoring physiological balance between the platelet and vessel.
Introduction
Diabetes mellitus is a chronic, progressive disease characterized by abnormal carbohydrate, lipid and protein metabolism. These abnormalities lead to the formation of glycation end products and the accumulation of reactive oxygen species resulting in endothelial dysfunction and platelet activation [1, 2]. The cytokines and eicosanoids synthesized by the impaired endothelium and activated platelets are involved in the development and progression of atherosclerosis [3]. Eicosanoids, which regulate cellular and vascular function, are generated from free arachidonic acid (AA) released from phospholipids (PLs) in the cell membrane by phospholipase A2 in the presence of ionized calcium, by cyclooxygenases (COXs), lipoxygenases (LOXs) and specific synthetases [4]. The sigma-1 receptor (S1R), which modulates cellular functions [5–7], is expressed on both endothelial cells [8] and platelets [9]. Beneficial effects of S1R agonists have already been reported in sepsis [10], ischemic-reperfusion injury [11], stroke [12] and cardiovascular disease [13]. The selective S1R agonist PRE-084 (2-(4-morpholino)ethyl-1-phenylcyclohexane-1-carboxylate) [14] can regulate cytokine production in stroke [15], reduce microglial activation in traumatic brain injury, and exert a protective effect in neurogenic inflammation [16] and endothelial barrier damage [17]. NE-100 (N,N-dipropyl-2-[4-methoxy-3- (2-phenylethoxy)-phenyl]-ethylamine monohydrochloride) [18], known as an S1R antagonist, blocks thoracic aortic vasodilation induced by a sigma-1 agonist (SA4503) [13].
In our previous in vitro studies we examined the effects of S1R modulation on platelet functions. We demonstrated that the S1R agonist PRE-084 enhanced both eicosanoid synthesis and ADP- or AA-induced aggregation of isolated platelets from healthy rats. In this study, we examined only the acute effect of PRE-084 [9]. In a separate, follow-up study, intraperitoneal sub-chronic in vivo PRE-084 treatment reduced the total quantity of ex vivo/in vitro COX-mediated AA metabolites in both healthy rat platelets and aorta, although the incubation mixture did not contain S1R ligand. In addition to the S1R agonist PRE-084, the effects of the antagonist NE-100 and a novel ligand, (S)-L1 (S-N-Benzyl-6,7-dimethoxy-1,2,3,4-tetrahydro-1-isoquinolineethanamine), on ex vivo AA metabolism in healthy rat platelets and aorta were also studied [19].
The ligand (S)-L1 was identified by a controlled virtual screening protocol based on its high binding affinity to S1R (Ki = 11±3 nM) and moderate sigma-1/sigma-2 receptor selectivity [20] and was chosen to investigate its effects on eicosanoid synthesis in platelets and aorta. The S1R ligands in the screening study all conformed to the early pharmacophore model [21], which consists of the basic amine site required to form an electrostatic bond with Glu172 of the S1R and the hydrophobic groups flanking it. The main difference between agonist and antagonist binding is the presence of an interaction of the ligand with the C-terminal helix of the S1R. The cyclohexane ring of PRE-084 is the only moiety that is able to interact with this helix [22]. The (S)-L1 and S1R antagonist NE-100 have very similar binding positions [19, 20].
In vivo sub-chronic treatment of healthy rats with (S)-L1 resulted in a similar but greater change in platelet AA metabolism than PRE-084, while NE-100 ligand induced an opposite alteration in platelet eicosanoid synthesis to both the ligands PRE-084 and (S)-L1 [19].
It is well established in the literature that S1R gene expression reduces the production of inflammatory cytokines [10], reactive oxygen species and advanced glycation end products [2, 3] as well as abnormal protein accumulation in diabetes [23] and protects against the development of diabetic complications [24]. Based on these observations and our previous studies, the aim of the present study was to investigate the effects of sub-chronic intraperitoneal-administered S1R ligands PRE-084, (S)-L1 and NE-100 on platelet and aortic eicosanoid synthesis in streptozotocin (STZ)-induced diabetic rats.
We hypothesized that (1) S1R gene (Sigmar1) expression increases in diabetic rat platelets ex vivo suggesting a compensatory role of S1R in diabetic metabolic alterations; (2) In vivoactivation of platelet and endothelial dysfunction can be confirmed even after one month of disease and detected by ex vivo studies of platelets and aorta in STZ-induced diabetic rats; (3) The S1R agonist PRE-084 and the antagonist NE-100 have opposite effects on the levels of Sigmar1 and COX (prostaglandin G/H synthase; Ptgs) gene mRNA in diabetic rat platelets, whereas the novel S1R ligand (S)-L1 acts as an antagonist under the present conditions due to its similar structure to NE-100; (4) S1R ligands administered sub-chronically in vivo are able to modulate ex vivo platelet and aortic AA metabolism, despite the absence of ligand in the incubation mixture; and (5) Abnormal AA metabolism in platelets and aorta of diabetic animals is modified by S1R ligands in such a way that the physiological balance between them is restored.
To confirm these hypotheses, we measured the serum level of the S1R ligands at the end of sub-chronic treatment and the mRNA levels of Sigmar1 and Ptgs genes in platelets and examined the eicosanoid synthesis in platelets and aorta ex vivo in diabetic rats treated with S1R ligands and compared the results to a vehicle-treated, healthy control group.
…
Results
All the results from the healthy, vehicle-treated rats, obtained in parallel with the diabetic animals, can be found in Váczi et al. (doi: 10.1016/j.ejphar.2022.174983) [19]. All data used in this manuscript have been uploaded to a data repository and made publicly available at link https://osf.io/5wgdb/files/osfstorage.
Physical and laboratory parameters of the animals
STZ-induced diabetic model.
As checked in the initial and the terminal phase, all the STZ-treated animals were hyperglycaemic. The fasting blood glucose levels were ~5 times higher than the basal glucose levels (Table 1) or those of the vehicle treated healthy rats tested in parallel with the present experiment [19]. In addition to hyperglycaemia, the development of diabetes was confirmed by the fact that the body weight gain of all the STZ-treated animals was significantly lower in the terminal phase as compared to that of the vehicle-treated, healthy rats. There were no significant differences between terminal blood glucose levels and body weight in diabetic animals receiving different treatments (vehicle, PRE-084, (S)-L1 and NE-100). The platelet counts were also in the same range, except for the NE-100 treatment group, in which an elevated terminal platelet count was measured (Table 1).

Body weight, fasting serum glucose levels and platelet number of diabetic rats.
At the end of the in vivo study (terminal phase), the levels of total cholesterol (Chol), alanine aminotransferase (ALT) and blood urea nitrogen (BUN) were determined from the pooled plasma samples (Fig 1).
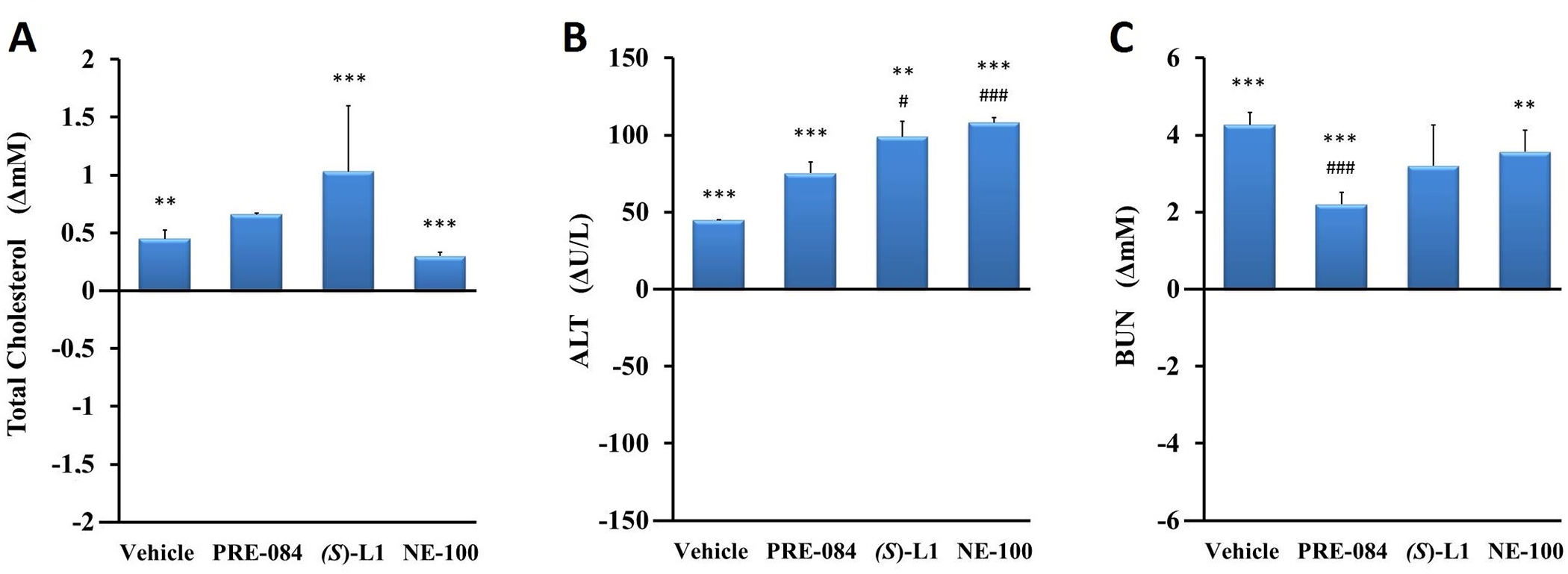
Laboratory parameters of rats at the end (terminal phase) of the in vivostudy.
Plasma total cholesterol (A), alanine aminotransferase (ALT) (B), and blood urea nitrogen (BUN) (C) levels in diabetic vehicle, PRE-084, (S)-L1 or NE-100-treated rats are shown as compared to the vehicle-treated, healthy animals. The zero line in the diagram shows the mean of the plasma concentrations in the vehicle-treated, healthy rats, while the columns in the figure represent the delta mean±SD values for the diabetic treatment groups; n = 3 samples pooled from nine rats per group; Welch’s ANOVA, Dunnett’s T3 test, **p<0.03, ***p<0.01 compared to the vehicle-treated, healthy rat group; #p<0.05, ###p<0.01 compared to the vehicle-treated, diabetic rat group.
The serum Chol, ALT and BUN levels in the STZ-induced diabetic animals treated with vehicle were higher (Chol by 21%; ALT by 120%; BUN by 63%) than those of the healthy rats studied in parallel with the present experiment. PRE-084 and NE-100 significantly elevated serum Chol and BUN in the diabetic rats compared to the vehicle-treated, healthy animals. In the diabetic rats, all three S1R ligands further enhanced the STZ-induced increase in plasma ALT levels compared to the vehicle-treated, healthy animals. None of the S1R ligands tested significantly altered the rise in serum Chol levels induced by STZ treatment. (S)-L1 and NE-100 further enhanced the growth in ALT levels observed in the vehicle-treated, diabetic rats. The smallest growth in ALT was detected when PRE-084 was used. An STZ-induced rise in BUN levels was only attenuated by PRE-084 (Fig 1).
Concentration of S1R ligands in rat plasma
In our preliminary experiments, we demonstrated that i.p. administered S1R ligands appear in the circulation within one hour, but their plasma levels are at the limit of detection after 20 hours [19]. Plasma concentrations of S1R ligands were determined in all diabetic rats 20 hours after the last ligand injection, immediately before the ex vivo studies. In the diabetic rats, the plasma concentration of PRE-084 was below the quantification limit, that of (S)-L1 was 10.99±1.06 mM, and that of NE-100 was 5.31±3.14 mM (data mean±SD; n = 9 rats per group). PRE-084 was eliminated from the circulation the fastest of all the Sigma-1 ligands we tested, while (S)-L1 was eliminated the slowest [19].
Effects of S1R ligands on the level of Sigmar1 and Ptgs1 transcripts in the diabetic rat platelets
The mRNA levels of Sigmar1 were detected in rat platelet samples using RT-qPCR (Fig 2).
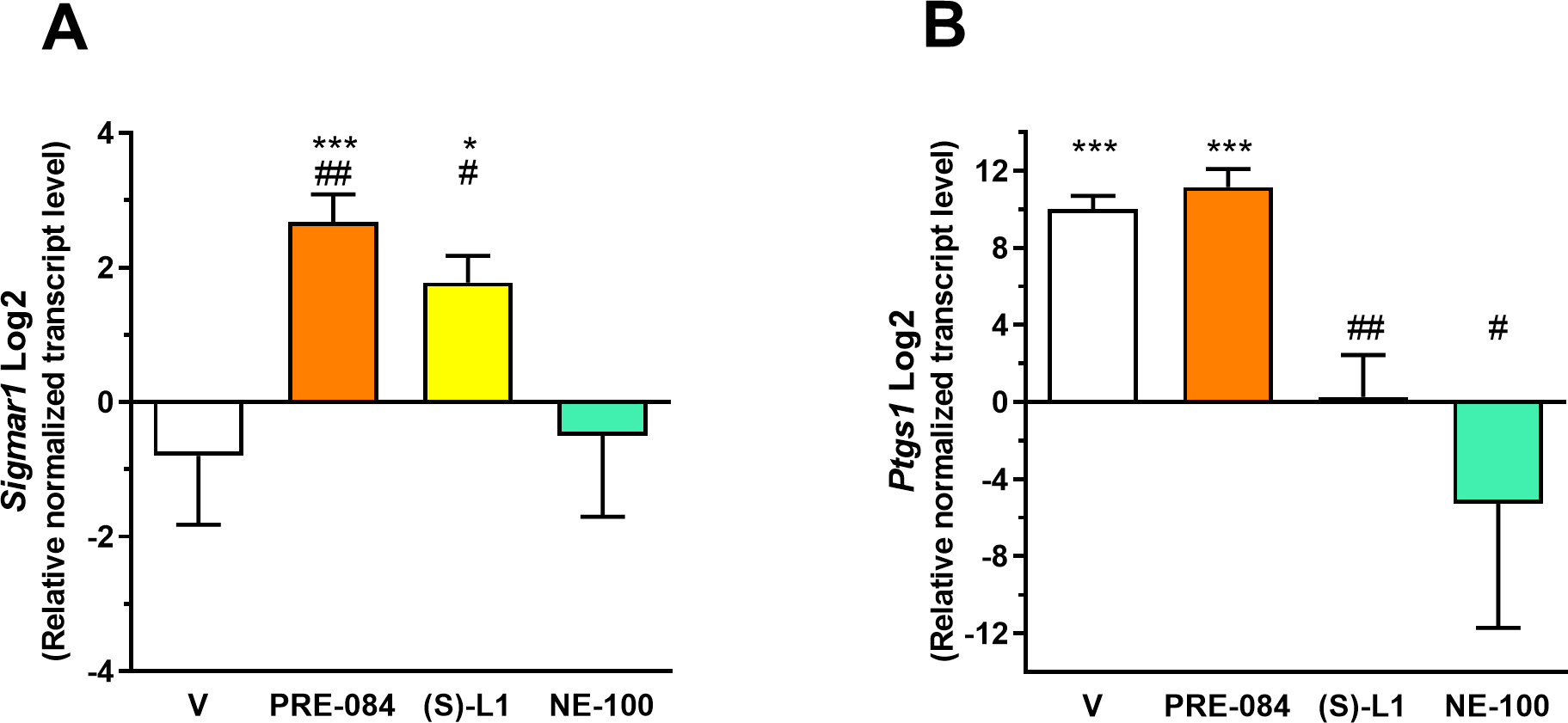
Relative transcript levels of Sigmar1 and COX-1 (Ptgs1) genes in platelet samples from diabetic rats treated with S1R ligands normalized to the healthy control group.
The diabetic rats were treated with PRE-084, (S)-L1, NE-100 or saline solution (V, vehicle) for seven days. The transcript levels of Sigmar1 (A) and Ptgs1 (B) genes in the rat platelet samples were first normalized to their own endogenous control mRNA levels (Gapdh) and then to the similarly normalized mRNA levels in the vehicle-treated, healthy animal group. Fold changes were calculated using the 2−ΔΔCt formula. Mean±SD, n = 4 representing nine rats per each group, Welch’s ANOVA, Dunnett’s T3 test, *p<0.05, ***p<0.001 compared to the vehicle-treated, healthy group; #p<0.05, ##p<0.01 compared to the vehicle-treated, diabetic group.
A low concentration level of the Sigmar1 mRNA was detected in the rat platelets from the vehicle-treated, diabetic animals (Fig 2A), similarly to the platelet samples from the healthy rats as published in our previous study [19]. Daily administration of PRE-084 and (S)-L1 for one week to diabetic rats elevated Sigmar1 transcript levels as compared to samples from the vehicle-treated, diabetic or healthy rats (Fig 2A) [19] rats. The Sigmar1 mRNA level was lower in the NE-100-treated diabetic group compared to those in the PRE-084 and (S)-L1-treated samples but did not differ significantly from the vehicle-treated, diabetic (Fig 2A). or healthy groups.
The level of Ptgs1 transcript was significantly higher in the platelets of the vehicle-treated, diabetic rats compared to those of the vehicle-treated, healthy animals (Fig 2B). In the diabetic rats, PRE-084 treatment did not change Ptgs1 mRNA levels as compared to samples from the vehicle-treated, diabetic animals. The Ptgs1 mRNA level was lower in the (S)-L1 group than in the vehicle or PRE-084-treated diabetic rat group, but did not differ significantly from the vehicle-treated, healthy group (Fig 2B). NE-100 treatment in diabetic rats significantly decreased the platelet Ptgs1 level as compared to the vehicle-treated, diabetic group (Fig 2B). Interestingly, the effect of (S)-L1 was different in the case of Sigmar1 (Fig 2A) and Ptgs1 (Fig 2B) mRNA levels: an effect similar to the S1R agonist PRE-084 was observed for Sigmar1mRNA level (Fig 2A), while a trend similar to the S1R antagonist NE-100 was seen for the Ptgs1 mRNA concentration in the rat platelets (Fig 2B). Ptgs2 mRNA was not detected in the platelet samples by RT-qPCR using 40 cycles in the diabetic or healthy rats.
Analysis of ex vivo AA metabolism in diabetic rats
Effects of S1R ligands on the ex vivo eicosanoid synthesis of platelets.
Platelet eicosanoid synthesis from radioactive AA substrate. In the platelets, one month after the development of STZ-induced diabetes, neither the total quantity of COX metabolites (the sum of 6-keto prostaglandin (PG) F1α (6-k-PGF1α), which is a stable metabolite of prostacyclin; PGF2α; PGE2; PGD2; thromboxane B2 (TxB2), which is a stable metabolite of TxA2 and 12-L-hydroxy-5,8,10-heptadecatrienoic acid), nor the total quantity of eicosanoids formed by the LOX pathway was altered (Fig 3A) compared to healthy rats.
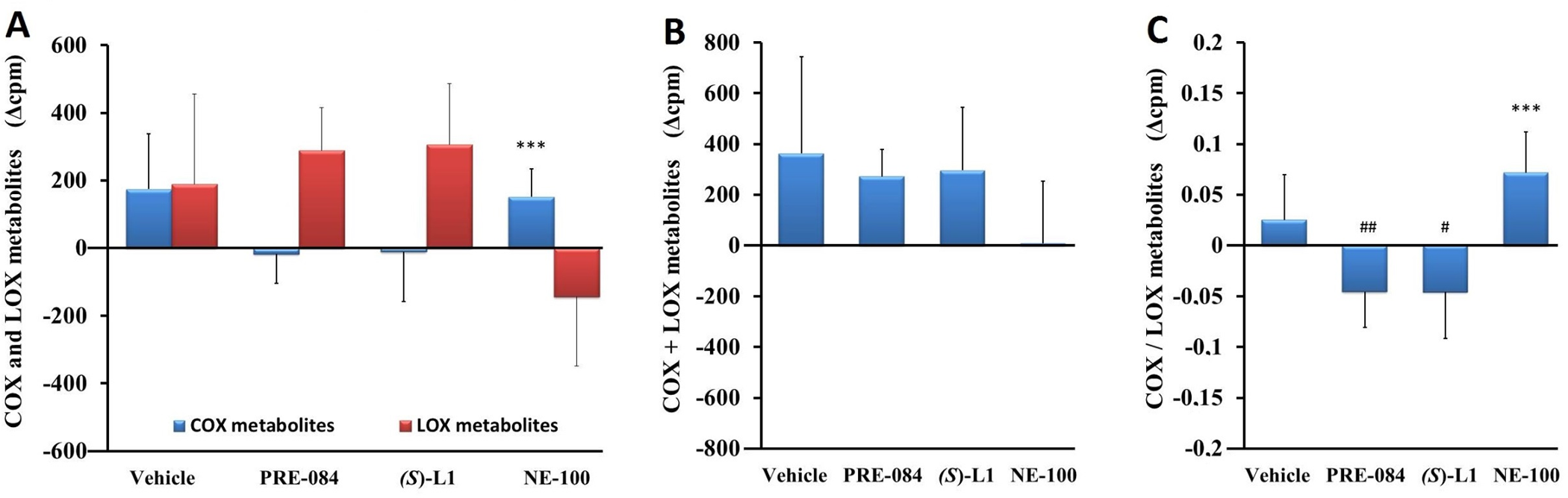
Arachidonic acid metabolism in rat platelets.
COX and LOX AA metabolites (A), total quantity of AA metabolites (COX+LOX) (B) and the ratio of COX to LOX AA metabolites (COX/LOX) (C) in the platelets from the diabetic vehicle, PRE-084, (S)-L1 or NE-100-treated rats are shown as compared to the vehicle-treated, healthy animals. The zero line in the diagram shows the mean for the isotope activity of the vehicle-treated, healthy rat platelets, while the columns in the figure represent the delta mean±SD value of the diabetic treatment group; n = 9 samples per group; Welch’s ANOVA, Dunnett’s T3 test, ***p<0.01 compared to the vehicle-treated, healthy rats group; #p<0.05, ##p<0.03 compared to the vehicle-treated, diabetic rat group; cpm: count per minute; COX: total quantity of cyclooxygenase metabolites synthesized from AA in platelets; LOX: total quantity of lipoxygenase metabolites synthesized from AA in platelets.
In the diabetic rat platelets, the total quantity of the COX metabolites was significantly eleveted by NE-100 compared to the vehicle-treated, healthy rat platelets. The total quantity of the AA metabolites (COX+LOX) in the diabetic platelets was not significantly modified by the S1R ligands compared to the vehicle-treated, healthy and diabetic samples (Fig 3B). Although there was no detectable change in platelet COX/LOX ratio one month after the STZ-induced development of diabetes, treatment with PRE-084 or (S)-L1 significantly reduced it compared to the vehicle-treated, diabetic samples. Meanwhile, NE-100 increased the COX/LOX ratio of the diabetic platelets as compared to the vehicle-treated, healthy rat platelets (Fig 3C).
In the platelets from the vehicle-treated, diabetic rats, the synthesis of TxB2 was significantly decreased (Fig 4A), while the production of PGD2 (Fig 4B), vasodilator and platelet anti-aggregator product was not significantly changed compared to the healthy animals (Fig 4).
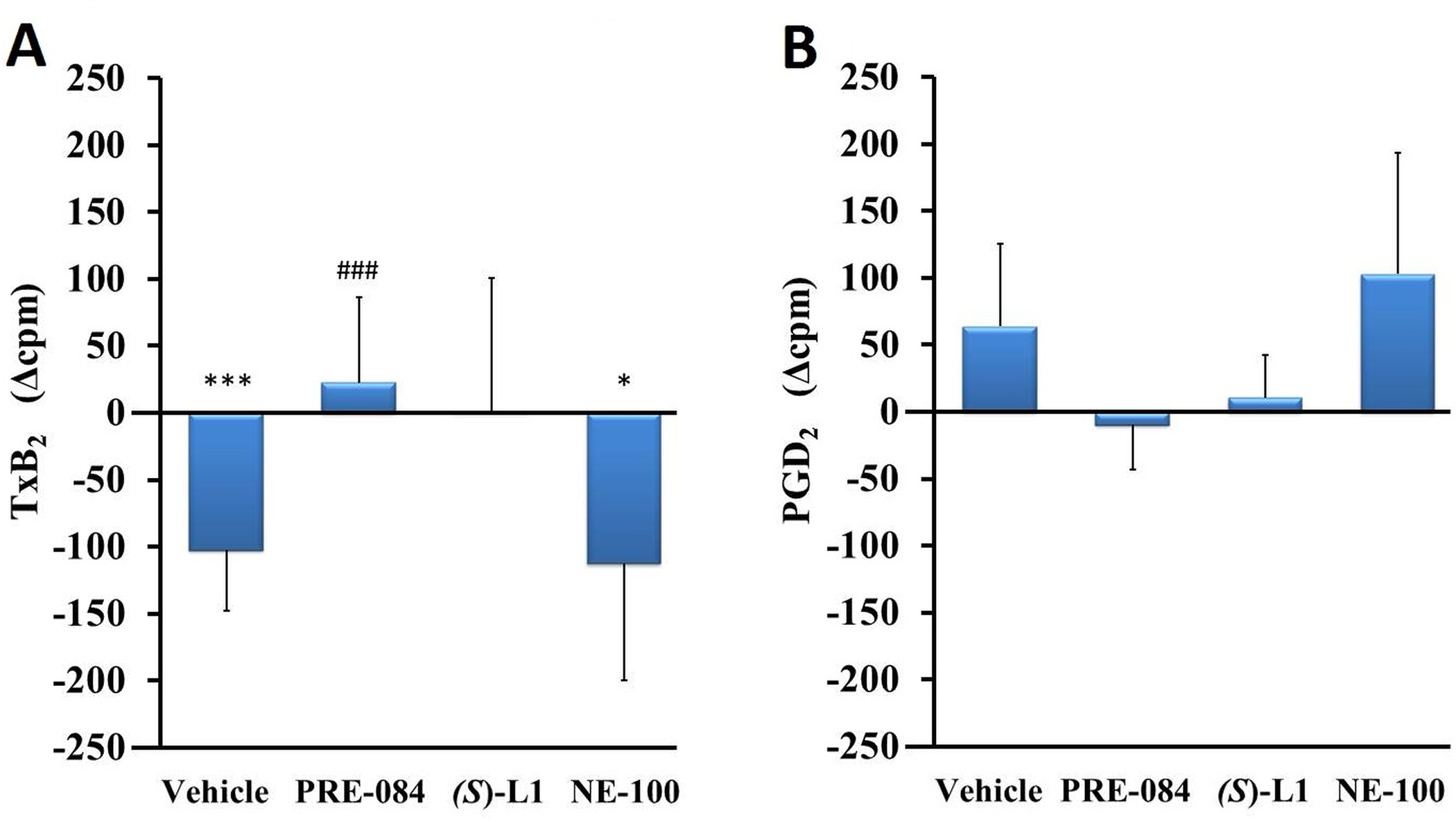
The effects of S1R ligands on the different COX metabolites in rat platelets.
TxB2 (A) and PGD2 (B) COX metabolites in the platelets from diabetic vehicle, PRE-084, (S)-L1 or NE-100-treated rats are shown as compared to the vehicle-treated, healthy animals. The zero line in the diagram shows the mean for the isotope activity of the vehicle-treated, healthy rat platelets, while the columns in the figure represent the delta mean±SD values for the diabetic treatment groups; n = 9 samples per group; Welch’s ANOVA, Dunnett’s T3 test, *p<0.05, ***p<0.01 compared to the vehicle-treated, healthy rat group; ###p<0.01 compared to the vehicle-treated, diabetic rat group; cpm: count per minute; TxB2: thromboxane B2; PGD2: prostaglandin D2.
PRE-084 elevated the synthesis of TxB2 in diabetic platelets compared to the vehicle treated, diabetic rat platelets, while NE-100 reduced it compared to the vehicle-treated, healthy rat platelets (Fig 4A).
In the vehicle-treated, diabetic rat platelets, the synthesis of the vasoconstrictor and platelet aggregator COX metabolites (CON: the sum of PGF2α and TxB2) was not changed significantly, while the quantity of vasodilator and anti-aggregator COX products (DIL: sum of PGE2 and PGD2) was increased (Fig 5A) compared to the healthy rat platelets.
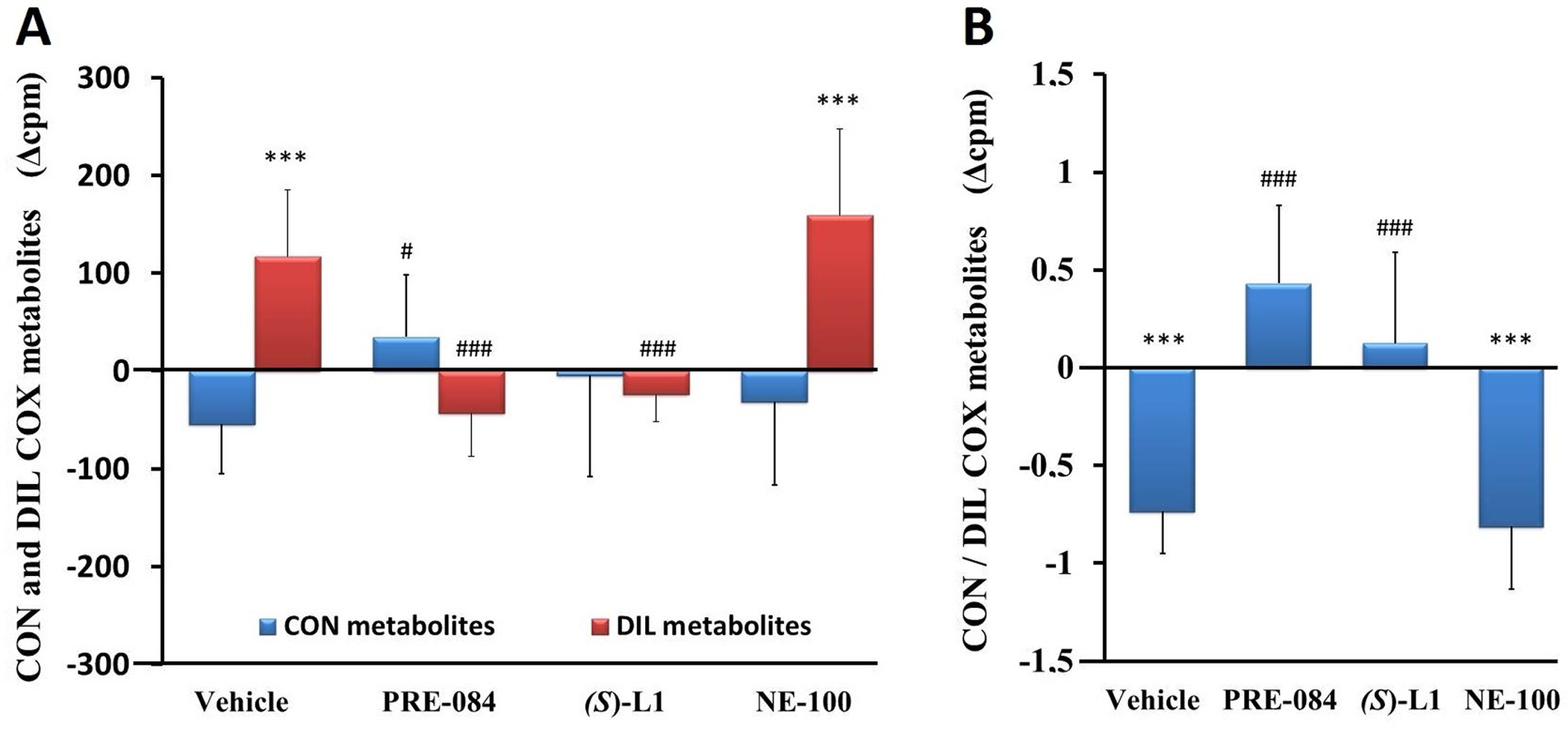
Vasoconstrictor, platelet aggregator (CON) and vasodilator, platelet anti-aggregator (DIL) COX metabolites of rat platelets.
CON COX and DIL COX AA metabolites (A) and the ratio of CON and DIL COX metabolites (CON/DIL) (B) in the platelets from the diabetic vehicle, PRE-084, (S)-L1, or NE-100-treated rats are shown as compared to the vehicle-treated, healthy animals. The zero line in the diagram shows the mean for the isotope activity of the vehicle-treated, healthy rat platelets, while the columns in the figure represent the delta mean±SD values for the diabetic treatment groups; n = 9 samples per group; Welch’s ANOVA, Dunnett’s T3 test, ***p<0.01 compared to the vehicle-treated, healthy rat group; #p<0.05, ###p<0.01 compared to the vehicle-treated, diabetic rat group; cpm: count per minute; CON: the sum of PGF2α and TxB2; DIL: the sum of PGE2 and PGD2.
The quantity of DIL COX metabolites in the diabetic platelets treated with NE-100 was significantly higher (Fig 5A) and consequently the ratio of CON to DIL COX metabolites was lower than in the healthy platelets from the vehicle-treated rats (Fig 5B). The quantity of the CON COX metabolites was raised in the diabetic platelets by the ligand PRE-084 as compared to the vehicle-treated, diabetic platelets. The synthesis of the DIL COX metabolites was reduced not only by PRE-084, but also by (S)-L1 compared to the vehicle-treated, diabetic group (Fig 5A). The CON/DIL ratio was significantly lower in the vehicle-treated, diabetic rat platelets than in the vehicle-treated, healthy ones (Fig 5B). In the platelets in the diabetic rats, an increase in CON/DIL ratio was observed in both the PRE-084 and (S)-L1-treated groups compared to the vehicle-treated, diabetic rat platelets (Fig 5B)
COX enzyme levels in rat platelets as determined by ELISA. The concentration of constitutive COX-1 in the vehicle-treated, diabetic rat platelets was not different, whereas the concentration of inducible COX-2 was significantly higher than in the vehicle-treated, healthy platelets (Fig 6A).
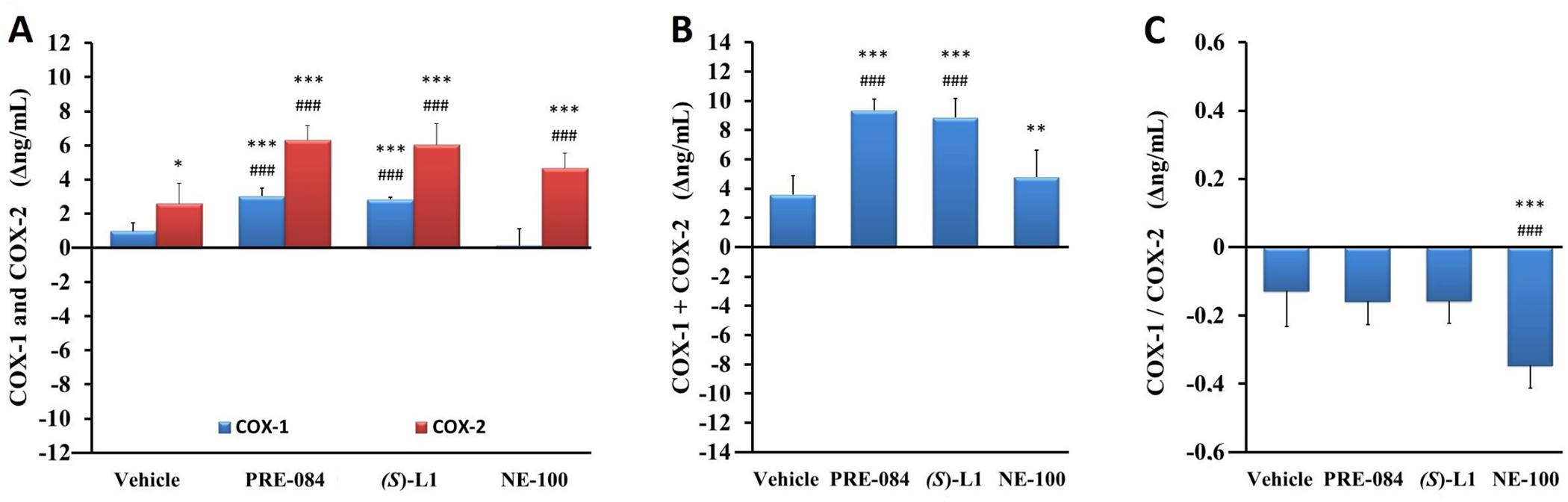
Effect of S1R ligands on the quantity of COX-1 and COX-2 enzymes in rat platelets as determined by ELISA.
Level of COX-1 and COX-2 enzymes (A), the total quantity of COX enzymes (COX-1+COX-2) (B) and the ratio of COX-1 to COX-2 (COX-1/COX-2) (C) in the platelets from the diabetic vehicle, PRE-084, (S)-L1 or NE-100-treated rats are shown as compared to the vehicle-treated, healthy animals. The zero line in the diagram shows the mean for the concentration of COX enzymes of the vehicle-treated, healthy rat platelets, while the columns in the figure represent the delta mean±SD values for the diabetic treatment groups; n = 9 samples per group; Welch’s ANOVA, Dunnett’s T3 test, **p<0.02, ***p<0.01 compared to the vehicle-treated, healthy rat group; ###p<0.01 compared to the vehicle-treated, diabetic rat group; COX-1: Type 1, constitutive cyclooxygenase; COX-2: Type 2, inducible cyclooxygenase.
In the diabetic rat platelets, the concentration of COX-1 was increased by the ligands PRE-084 and (S)-L1, while the COX-2 level was raised by all three S1R ligands as compared to both the vehicle-treated, healthy and diabetic rats (Fig 6A). The total quantity of COX enzymes (COX-1+COX-2), was also elevated by all three S1R ligands, but in the case of the NE-100 ligand this was due to rise in COX-2 levels (Fig 6B). This is supported by a decrease in the COX-1/COX-2 ratio of the rat platelets treated with the NE-100 ligand compared to the vehicle-treated, healthy and diabetic samples (Fig 6C).
Effects of S1R ligands on the ex vivo eicosanoid synthesis of the diabetic rat abdominal aorta.
Aortic eicosanoid synthesis from radioactive AA substrate. The total quantity of metabolites synthesized from AA by COX or LOX enzymes in the aorta samples of the vehicle-treated, diabetic animals did not differ significantly from that of the vehicle-treated, healthy rats (Fig 7A).
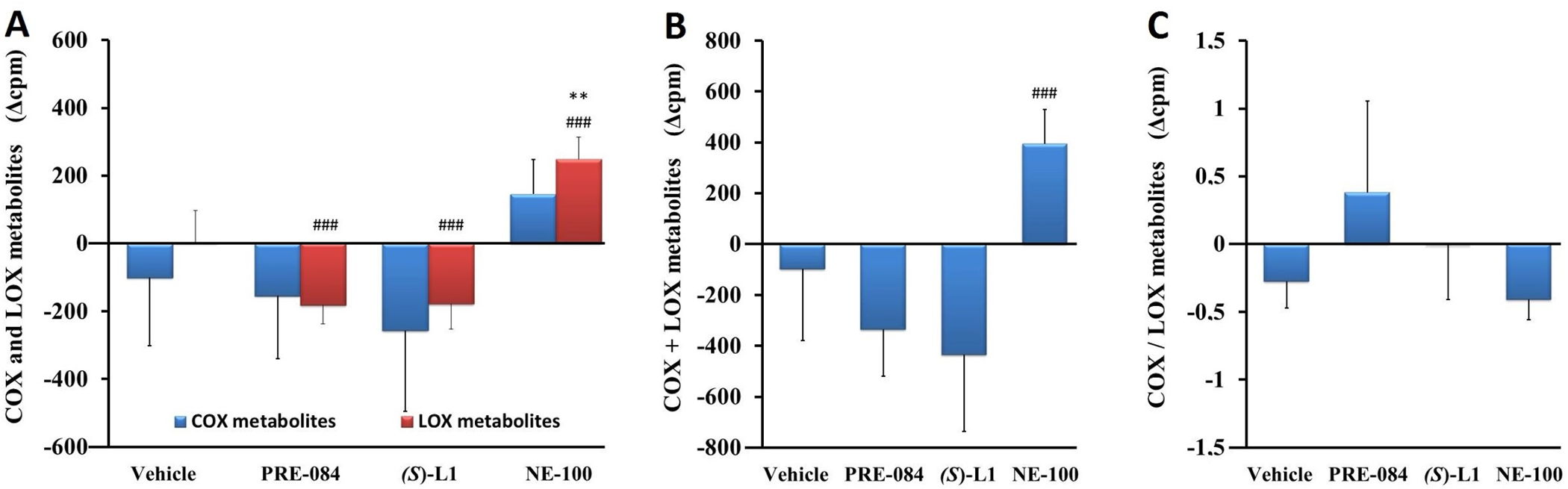
Arachidonic acid metabolism in rat abdominal aorta.
COX and LOX AA metabolites (A), total quantity of AA metabolites (COX+LOX) (B), and the ratio of COX to LOX AA metabolites (COX/LOX) (C) synthesized in the aorta of the diabetic vehicle, PRE-084, (S)-L1 or NE-100-treated rats are shown as compared to the vehicle-treated, healthy animals. The zero line in the diagram shows the mean for the isotope activity of the vehicle-treated, healthy rat aorta, while the columns in the figure represent the delta mean±SD value of the diabetic treatment groups; n = 9 samples per group; Welch’s ANOVA, Dunnett’s T3 test, **p<0.03 compared to the vehicle-treated, healthy rat group; ###p<0.01 compared to the vehicle-treated, diabetic rat group; cpm: count per minute; COX: total quantity of cyclooxygenase metabolites synthesized from AA in abdominal aorta; LOX: total quantity of lipoxygenase metabolites synthesized from AA in abdominal aorta.
The abdominal aortic AA metabolism (COX+LOX metabolites) (Fig 7B) and COX/LOX product ratio of the vehicle-treated, diabetic rat (Fig 7C) did not change compared to the vehicle-treated, healthy animal. The synthesis of LOX metabolites was significantly increased in the NE-100-treated, diabetic rat abdominal aorta compared to the vehicle-treated, healthy or diabetic one. PRE-084 and (S)-L1 administration reduced the production of LOX metabolites, without modifying the COX pathway in the diabetic aorta compared to the vehicle-treated, diabetic one (Fig 7A). The quantity of AA metabolites (COX+LOX) in the diabetic rat abdominal aorta was raised in the NE-100-treated group compared to the vehicle-treated, diabetic animals (Fig 7B). The COX/LOX metabolite ratio in the diabetic rat aorta was not modified by any of the S1R ligands compared to the vehicle-treated, healthy or diabetic rats (Fig 7C).
The synthesis of the vasoconstrictor TxB2 was not changed (Fig 8A), while the production of 6-k-PGF1α, the main vasodilator and platelet anti-aggregator product of the aorta, was decreased significantly in the vehicle-treated, diabetic rat aorta (Fig 8B) compared to the healthy animals.
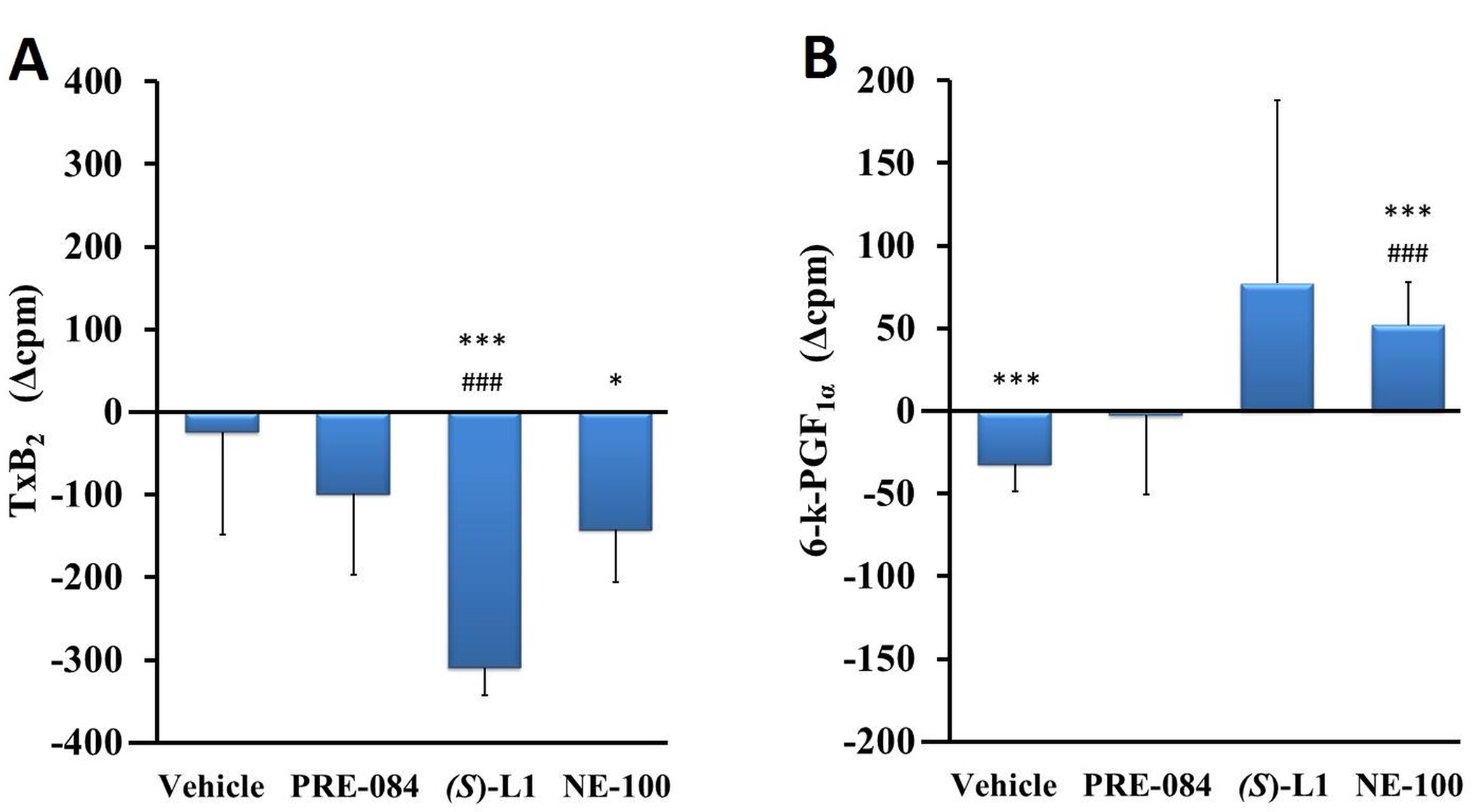
The effects of S1R ligands on the different COX metabolites in the rat aorta.
TxB2 (A) and 6-k-PGF1α (B) COX metabolites synthesized in the aorta of diabetic vehicle, PRE-084, (S)-L1 or NE-100-treated rats are shown as compared to the vehicle-treated, healthy animals. The zero line in the diagram shows the mean for the isotope activity of the vehicle-treated, healthy, rat aorta, while the columns in the figure represent the delta mean±SD values for the diabetic treatment groups; n = 9 samples per group; Welch’s ANOVA, Dunnett’s T3 test, *p<0.05, ***p<0.01 compared to the vehicle-treated, healthy rat group; ###p<0.01 compared to the vehicle-treated, diabetic rat-group; cpm: count per minute; TxB2: thromboxane B2; 6-k-PGF1α: 6-keto prostaglandin PGF1α.
(S)-L1 induced the reduction of TxB2 synthesis in the diabetic aorta compared to the vehicle-treated, healthy and diabetic ones (Fig 8A). The NE-100 ligand had opposite effects on these eicosanoids in the diabetic rat abdominal aorta: it reduced the production of TxB2 compared to the vehicle-treated, healthy rats (Fig 8A) and increased the synthesis of 6-k-PGF1α compared to the vehicle-treated, healthy and diabetic rats (Fig 8B).
No alterations in CON COX and DIL COX metabolite were detected in the abdominal aorta of the vehicle-treated, diabetic rats (Fig 9A) compared to the aorta of the vehicle-treated, healthy animals.
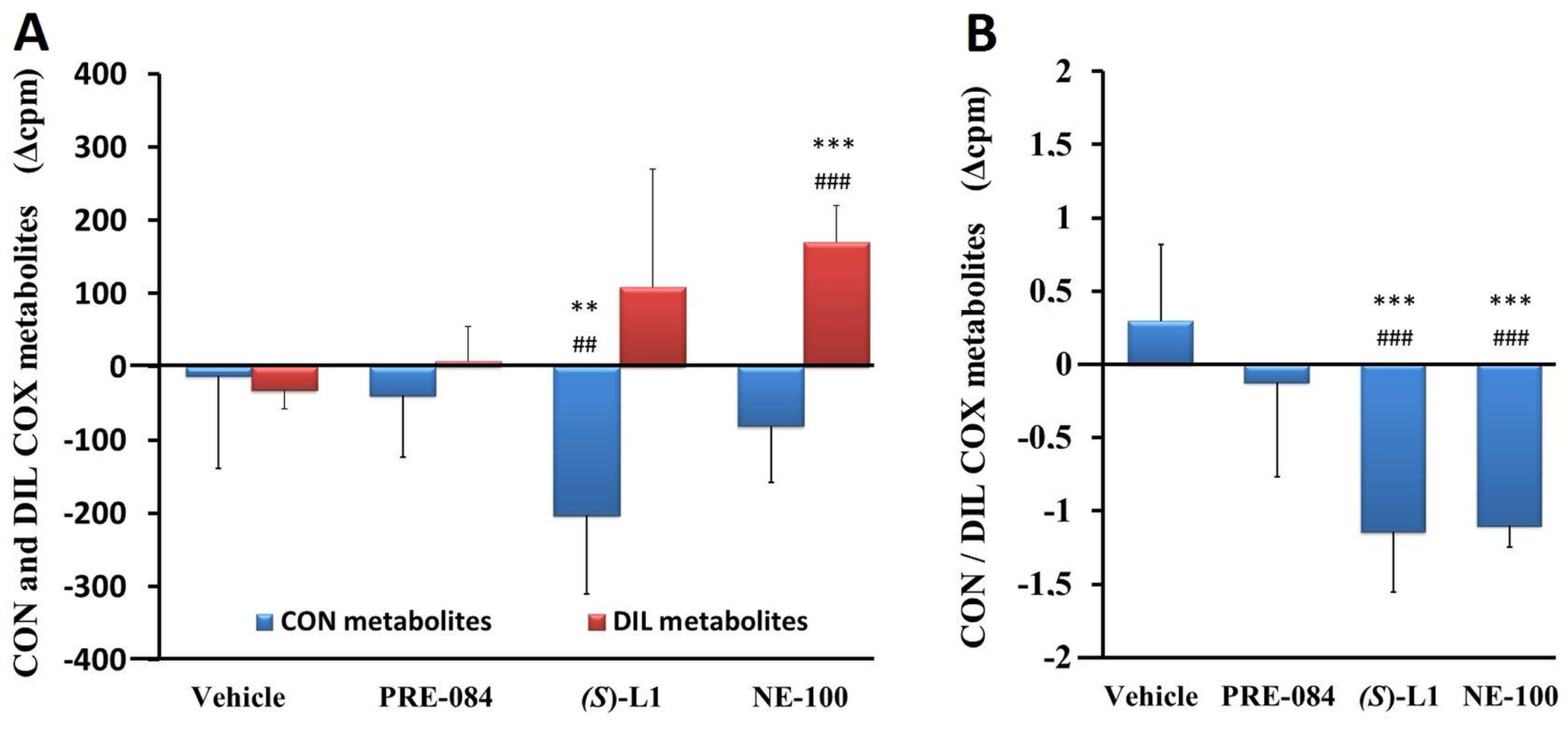
Vasoconstrictor, platelet aggregator (CON) and vasodilator, platelet anti-aggregator (DIL) COX metabolites of rat abdominal aorta.
CON COX and DIL COX metabolites (A) and the ratio of CON to DIL COX metabolites (CON/DIL) (B) synthesized in the aorta of the diabetic vehicle, PRE-084, (S)-L1 or NE-100-treated rats are shown as compared to the vehicle-treated healthy animals. The zero line in the diagram shows the mean for the isotope activity of the vehicle-treated, healthy rat aorta, while the columns in the figure represent the delta mean±SD values for the diabetic treatment groups; n = 9 samples per group; Welch’s ANOVA, Dunnett’s T3 test, **p<0.03, ***p<0.01 compared to the vehicle-treated, healthy rat group; ##p<0.03, ###p<0.01 compared to the vehicle-treated, diabetic rat group; cpm: count per minute; CON: the sum of PGF2α and TxB2; DIL: the sum of 6-k-PGF1α (that is a stable metabolite of prostacyclin), PGE2 and PGD2.
The production of CON COX metabolites of the diabetic rat aorta was significantly reduced by (S)-L1 (Fig 9A) compared to the vehicle-treated, healthy and diabetic groups. The synthesis of DIL COX metabolites in the diabetic rat abdominal aorta was stimulated by NE-100 compared to the vehicle-treated, healthy and diabetic samples (Fig 9A). Both the (S)-L1 and the NE-100 ligands significantly decreased the CON/DIL ratio in the diabetic rat abdominal aorta ring compared to the vehicle-treated, healthy and diabetic samples (Fig 9B).
COX enzyme levels in the abdominal rat aorta measured by ELISA. The concentration of COX-2 in the aortas of both healthy and diabetic vehicle-treated rats was higher than that of COX-1. Neither the concentration of COX-1 or COX-2 (Fig 10A) nor their sum (COX-1+COX-2) (Fig 10B) in the aorta of the vehicle-treated, diabetic rats showed significant differences compared to vehicle-treated, healthy animals.
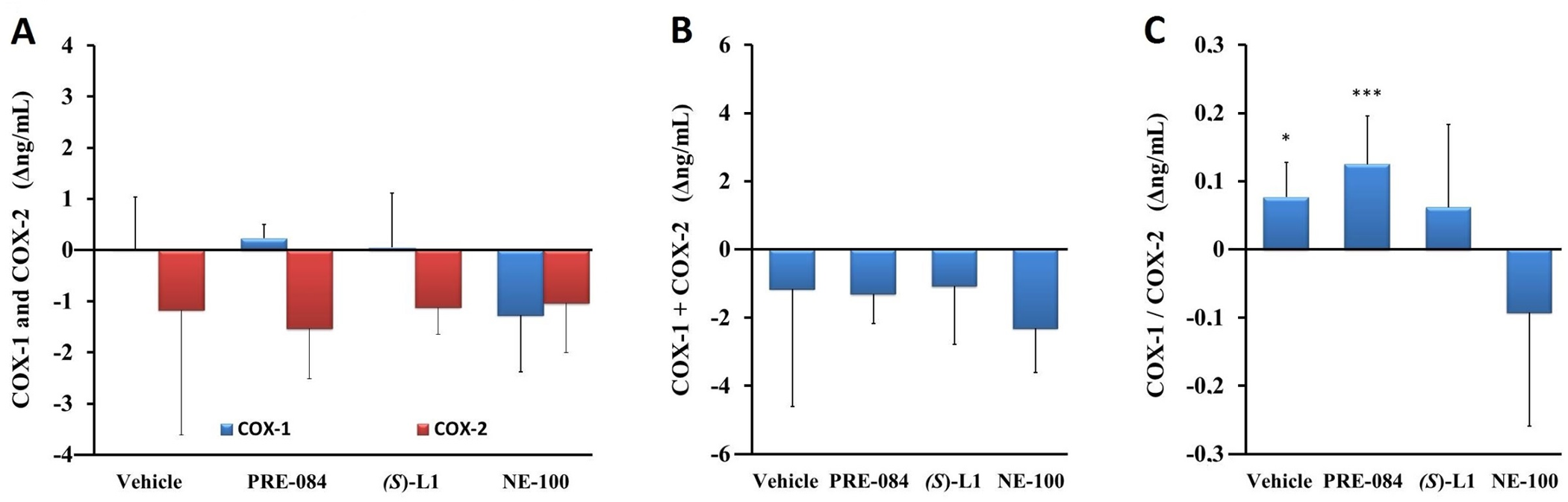
Effect of S1R ligands on levels of COX-1 and COX-2 enzymes determined by ELISA in rat abdominal aorta.
Enzyme concentrations of COX-1 and COX-2 (A), the total quantity of COX enzymes (COX-1+COX-2) (B) and the ratio of COX-1 to COX-2 (COX-1/COX-2) (C) in the aorta of the diabetic vehicle, PRE-084, (S)-L1 or NE-100-treated rats are shown as compared to the vehicle-treated, healthy animals. The zero line in the diagram shows the mean for the enzyme concentration of the vehicle-treated, healthy rat aorta, while the columns in the figure represent the delta mean±SD values for the diabetic treatment groups; n = 9 samples per group; Welch’s ANOVA, Dunnett’s T3 test, *p<0.05, ***p<0.01 compared to the vehicle-treated healthy rat group; #p<0.05 compared to the vehicle-treated diabetic rat group; COX-1: Type 1 cyclooxygenase; COX-2: Type 2 cyclooxygenase.
In the aorta of diabetic rats, the concentrations of these parameters were not altered by any of the S1R ligands we tested (Fig 10A, 10B). However, the COX-1/COX-2 ratio in the aorta of diabetic rats treated with vehicle or PRE-084 was significantly higher than in healthy rats treated with vehicle (Fig 10C).
Discussion
In the present study, we investigated the effects of sub-chronic, in vivo administered S1R ligands on ex vivo/in vitro eicosanoid synthesis of STZ-induced diabetic rat platelets and abdominal aorta. We selected STZ-induced diabetes mellitus as a reliable, reproducible model with chronic inflammation and oxidative stress, in which the development and time course of complications can be easily monitored [35]. STZ contains glucose and N-methyl-N-nitrosocarbamide groups [36]. Its glucose component binding to glucose transporter-2 (GLUT-2) promotes the entry of STZ into pancreatic, liver and renal tubule epithelial cells with GLUT-2 [37, 38]. The N-methyl-N-nitrosourea constituent of STZ thus transported into cells induces DNA methylation [39], alkylation [40] and oxidation [41]. These processes induce cell apoptosis, leading to the development of diabetes mellitus and liver and kidney damage.
In the present study, the development of STZ-induced diabetes mellitus was supported by elevated serum glucose level (hyperglycaemia) and a reduced rate of body weight gain in the rats compared to the physiological parameters. None of the ligands significantly altered the STZ-induced increase in serum glucose concentration and decreased body weight gain (Table 1), indicating they were unable to restore the STZ-induced metabolic changes. Consistent with our previous study [42], we found elevated serum cholesterol, ALT and urea levels in the STZ-induced diabetic animals, which may be explained by the hepatic and renal toxic effects of STZ [37, 38]. The STZ-induced rise in serum ALT concentration was further boosted by the S1R ligands, although the growth was not significant for PRE-084 (Fig 1). These results suggest the metabolism and/or elimination of ligands in the liver or kidney [43]. The platelet count below the reference value [44], already observed in our previous study, can be explained by reduced thrombopoietin production due to the liver and kidney damage associated with STZ-induced diabetes [42]. However, in the present study, we only observed a trend towards a decrease in platelet counts in the STZ-treated animals compared to the vehicle-treated, healthy rat group, which was normalized by PRE-084 and NE-100. The slightly reduced platelet number in diabetic rats does not influence the results of the present study, as eicosanoid synthesis was monitored at a standard platelet count (Table 1).
Ha et al. (2012) reported a protective role of S1R against pathological changes in STZ-induced diabetes mellitus [24]. Since both platelets and endothelial cells are involved in the development of diabetic complications [45], we hypothesized that S1R ligands could modulate the function of both cell types. We selected PRE-084, NE-100 and (S)-L1 from among the S1R ligands based on their binding strength to the receptor and their binding site in the S1R binding pocket as determined in our previous experiments [19]. Based on platelet lifespan, the duration of in vivo treatment was one week [46, 47]. Preliminary time-dependent studies of the serum levels of i.p. administered S1R ligands demonstrated that all ligands entered the circulation. However, their serum concentrations were only at the limit of detection 20 h after the last treatment; that is, it was possible to rule out the direct acute effect of the ligands during the ex vivo experiment. Thus, the ex vivo changes in platelet and abdominal aortic AA metabolism were attributed to the in vivo effects of S1R ligands on platelet and aortic function [19].
Sub-chronic, in vivo treatment with S1R ligands was also able to alter the AA metabolism of the platelets and aorta in the STZ-induced diabetic rats despite the absence of the ligands in the blood at the time of the ex vivo/in vitro study (Figs 3 and 7). S1R ligands can modulate the PL content of the cell membrane, the re- or de-acylation of PLs, and the amount of free AA released from PLs by phospholipases [4, 48, 49]. They may also affect the expression and/or activity of COX and LOX enzymes involved in AA metabolism. From the two COX isoforms, COX-1 is constitutively expressed in various tissues, but the expression of COX-2 is mostly inducible [50]. Although the platelets are anucleated cells, they can de novo synthesize COX-1 from the cytoplasmic mRNA that originates from the megakaryocyte [51]. In human platelets, Hu and co-workers [52] also detected COX-2 mRNA and protein, although at significantly lower levels than COX-1, consistent with our data. However, under the current experimental conditions, we did not detect Ptgs2 mRNA in the platelets from the healthy control [19] or diabetic rats with RT-qPCR. This may be explained by the fact that only limited amounts of megakaryocyte mRNA transcripts are available for protein synthesis (e.g. COX-2), and therefore, in vivo AA metabolism in platelets may lead to depletion of intracellular reserves (mRNA, enzyme pool and ☯Ca2+]i). Unchanged Sigmar1 and increased Ptgs1 transcript levels were detected in the platelets of the vehicle-treated, diabetic rats compared to the healthy ones. Based on these results, our hypothesis that platelets from the vehicle-treated, diabetic rats raised Sigmar1 transcript expression was not confirmed. However, under pathological conditions, S1R agonist PRE-084 with antioxidant properties [53] as well as the novel SR-1 ligand (S)-L1 were able to enhance S1R transcript level (Fig 2), which may have a protective role against oxidative stress in diabetes. NE-100 decreased Ptgs1 compared to the vehicle-treated, diabetic group. These results support our hypothesis that PRE-084 S1R agonist and NE-100 antagonist have opposite effects on platelet Sigmar1 and Ptgs1 transcript levels. The structural similarity between (S)-L1 and NE-100 is only reflected in the similar effect on platelet Ptgs1 levels, suggesting that the modulatory effect of S1R ligands is enzyme-, cell- and tissue-specific.
A rise in DIL COX products and a drop in the CON/DIL ratio were observed in the diabetic rat platelets (Fig 5), compared to the healthy animals, while a drop in 6-k-PGF1α synthesis was detected in aorta (Fig 8). These results support our hypothesis that platelet and endothelial cell activation in vivo can be detected ex vivo. In the platelets we observed a change opposite to that expected, which could be the result of a smaller storage pool of platelets (e.g. intracellular calcium ion) during in vivo platelet activation or a consequence of a compensatory mechanism that develops early in diabetes. In our previous ex vivo platelet aggregation study in STZ-induced diabetic rats, no increased platelet aggregation capacity was measured in diabetic rats either [42]. Consistent with the present data, decreased in vitro ADP- and AA-induced platelet aggregation was found in the STZ-induced diabetic rats compared to the healthy animals [54]. Paradoxically, these effects paralleled the high intrinsic hyperactivity of the platelets and might be explained by the time-dependent production of platelet aggregation-inhibiting factors.
Treatment with the ligands PRE-084 and (S)-L1 did not affect the total quantity of the radioactive eicosanoids (COX+LOX) synthesized by the platelets from the diabetic rats, but it lowered the COX/LOX ratio. These results suggest that PRE-084 and (S)-L1 induce a shift in platelet AA metabolism towards the LOX pathway without altering the amount of AA substrate released from PLs (Fig 3).
Although PRE-084 and (S)-L1 did not elevate COX-1 mRNA (Fig 2) in the diabetic platelets compared to the vehicle-treated diabetic platelets, they raised COX enzyme concentrations as determined with ELISA (Fig 6). While this did not induce a rise in the total quantity of COX pathway products (Fig 3), i.e. an increase in COX enzyme activity, it modulated the function of the enzymes involved in the synthesis of individual COX products to normalize the changes measured in diabetic platelets (Figs 4, 5). For example, in diabetic rats, a higher CON/DIL ratio was observed in the platelets in the PRE-084 and (S)-L1 treatment groups, indicating a predominance of CON COX metabolites (Fig 3) compared to platelets from the vehicle-treated, diabetic rats, which, in contrast, showed a reduced CON/DIL ratio compared to the vehicle-treated, healthy group. NE-100 did not affect the AA metabolism of platelets from diabetic animals compared to the vehicle-treated, diabetic group (Figs 3–6).
Although several safety procedures have been taken to prevent spontaneous aggregation of platelets during platelet isolation and in vitro studies, and no direct platelet aggregation was induced in ex vivo/in vitro studies, the possibility of activation of platelet intracellular signalling pathways cannot be completely excluded. EDTA used during platelet separation reduced the amount of extracellular calcium ion available to platelets, the calcium ion level was normalized by the tissue culture medium used in the ex vivo/in vitro study. In our ex vivo/in vitro study, as the tissue culture medium did not contain fibrinogen, platelet aggregation could not occur. We consider a limitation of our study that we have not studied a population of platelets at rest, but a population of platelets adapted to changes in the culture medium environment with physiological parameters. However, this does not make it impossible to compare the effects of S1R ligands ex vivo, as the ligand-treated and non-ligand-treated samples were tested in parallel, from the same platelet population.
The effects of S1R ligands on eicosanoid synthesis in abdominal aorta were very different from those observed in the platelets. Reduced synthesis of 6-k-PGF1α was detected in the aorta of diabetic rats (Fig 8). In the aorta, NE-100 was found to be the most potent S1R ligand in promoting the recovery of physiological status by enhancing 6-keto-PGF1α synthesis. The increase in the total quantity of AA metabolites (COX+LOX) in the NE-100 group (Fig 7) suggests that this is likely to be due not only to specific prostacyclin synthase, but also to the effect of higher AA substrate due to phospholipase activation. (S)-L1, in turn, increases the rate of vasodilator COX products (Fig 9) by decreasing the synthesis of vasoconstrictor, platelet aggregator eicosanoids compared to both vehicle-treated, diabetic and vehicle-treated, healthy rat aorta. In the aortic AA metabolism study, PRE-084 was the most ineffective ligand, not affecting the synthesis of COX pathway products compared to either vehicle-treated, healthy or vehicle-treated, diabetic rats. Based on these results, our hypothesis that S1R ligands applied sub-chronically in vivo can modulate both platelet and aortic AA metabolism ex vivo was confirmed. The AA metabolism of platelets and endothelial cells are known to differ significantly under healthy conditions. Platelets primarily synthesize the vasoconstrictor thromboxane, whereas endothelial cells primarily synthesize the vasodilator prostacyclin. The balance between these arachidonic acid metabolites plays a crucial role in maintaining normal local circulation [31, 39, 42]. Overall, S1R ligands have opposite effects on platelet and aortic AA metabolism in diabetic rats. Our hypothesis that S1R ligands modulate the pathological AA metabolism in diabetic rat platelets and aortic rings in such a way that the physiological balance between them is restored was confirmed.
Conclusion
In STZ-induced diabetic rats platelet TxB2 and aortic 6-k-PGF1α production dropped. Sub-chronic in vivo treatment of diabetic animals with the PRE-084 ligand enhanced CON COX and reduced DIL COX product formation, while (S)-L1 lowered the synthesis of DIL COX metabolites and promoted the recovery of physiological platelet function in diabetic rats. The S1R antagonist NE-100, on the other hand, produced no significant changes in the diabetic rat platelet AA metabolism. In contrast, (S)-L1 decreased the synthesis of CON COX metabolites, whereas NE-100 increased the quantity of aortic DIL COX products and promoted the recovery of diabetic endothelial dysfunction in the aorta. S1R agonist PRE-084 did not change aortic AA metabolism. The novel S1R ligand (S)-L1 had effects on eicosanoid synthesis in platelets similar to those of the agonist PRE-084 and on eicosanoid synthesis in aorta similar to those of the antagonist NE-100. Our results suggest that S1R ligands may play a role in the regulation of cellular functions and local circulation by affecting AA metabolism at transcription, translation and enzyme induction levels. In diabetes mellitus, the cell-specific effects of S1R ligands have a compensatory role and aid in restoring physiological balance between the platelet and vessel.